


多肽药物凭借其生物同源性、低毒性、高安全性、精准靶向性以及无体内蓄积等特性, 已成为现代医药领域的重要支柱[1]. 随着多肽和蛋白质合成及筛选技术的发展, 镜像多肽被开发出来, 并在疾病治疗、生物医药开发、生命起源以及镜像生命再造等多个领域表现出应用潜力, 受到科学家日益广泛的关注. 此外, 肽醇和多肽酰肼对于蛋白质合成、多肽药物开发等具有重要意 义[2]. 肽醇(Peptide alcohols)作为一类C末端经羟基(OH)修饰的功能化多肽, 因其独特的结构可调性和多样化的生物学功能[3], 在药物研发、生物技术及材料科学等领域展现显著潜力[4]. 羟基修饰不仅赋予肽醇分子更高的化学稳定性, 还可作为活性位点赋予其抗菌[5]、抗炎[6]、抗氧化[7]、免疫调节[8]及组织修复[9]等多重生物学活性. 同时维持了多肽固有的高选择性和低毒性特征, 成为疾病治疗的理想候选分子.
在药物合成领域, 肽醇的重要性同样不容忽视. 它们常作为肽醛的前体, 参与蛋白酶抑制剂和药物中间体的关键合成步骤[10]. 羟基的存在进一步优化了肽醇分子的生物活性和药代动力学特性, 例如增强其水溶性和代谢稳定性. 经典的肽醇药物包括奥曲肽(Octreo- tide)[11], 通过C末端羟基修饰显著提高化学稳定性和生物利用度, 成为治疗肢端肥大症和神经内分泌肿瘤的重磅药物, 并跻身全球多肽药物销售前列[12]. 抗菌肽醇Gramicidin A则凭借其独特的肽醇结构, 在膜靶向抗菌机制研究中占据重要地位[4]. 此外, 镇痛脑啡肽类似物Tyr-D-Ala-Gly-MePhe-Met(O)-ol[13]和Trichogin A IV[14]通过肽醇结构调控生物活性, 分别在神经调控和抗肿瘤领域展现出突破性应用前景.
通过这些经典案例可以看出, 肽醇凭借其多样化的功能和显著的药用潜力, 为多肽类药物的开发开辟了广阔的前景[15]. 未来, 随着合成技术的不断进步和生物活性研究的深入, 肽醇有望在更多疾病治疗领域发挥重要作用, 为人类健康提供更多创新解决方案. 然而, 尽管肽醇展现出巨大的应用价值, 目前已报道的有效合成肽醇的方法仍然有限, 严重制约了其大规模制备以及进一步的研究与应用. 现有合成方法往往面临效率低、成本高、环境友好性不足等问题, 难以满足工业化生产和科学研究的需求. 因此, 开发高效、绿色、经济的肽醇合成新方法, 已成为当前多肽药物研究领域亟待解决的关键问题之一.
肽醇的合成一般是通过修饰肽链的C末端来实 现[16](Scheme 1a). 然而在固相合成(SPPS)中, C端修饰非常困难[17]. 能直接用于肽醇合成商业化树脂较少, 通常需要对固相合成中的树脂进行C端修饰, 引入特殊的连接体(Linker), 然后与氨基醇反应接入第一个残基[11]. 这种方法已成为合成肽醇的一种流行方法, 然而, 羟基的锚定需要特定的连接体, 这类连接体普遍不稳定且与氨基醇的偶联效率较差, 限制了其商业化应用范围. Edwards等[18]报道了一种通过氨解反应即在裂解时引入氨基醇的方法, 然而, 氨解反应条件苛刻, 会产生各种副产物, 后续纯化困难. Tailhades等[4]报道了一种O—N酰基转移反应合成肽醇, 虽然这种方法适用于常规商业化树脂, 但其可行性受限于O—N酰基转移反应效率较低, 难以规模化放大. Crawhall和Elliott[19]报道了用LiAlH4和NaBH4还原树脂上的肽序得到氨基醇, 然而, 还原反应速率缓慢且条件苛刻, 得到复杂且不易纯化的产物. 为了解决这一问题, Kao等[20]最近报道了二氨基苯甲酰基(Dbz) 连接体的应用, 该连接体可满足还原性切割的要求, 能够通过NaBH4还原树脂上的肽序得到肽醇, 但此方法需对树脂进行复杂修饰, 操作繁杂且成本很高.
除了这些固相合成肽醇的方法, 目前未见在液相体系中合成肽醇的报道. 固相合成虽然是目前多肽化学合成的主流方法, 但也存在一些显著的局限性: 使用的树脂极其昂贵、需要大过量的氨基酸及偶联试剂投入反应以保证缩合反应的完成, 因此原子经济性极差[21]; 需要消耗大量的有机溶剂(DMF)洗涤树脂, 有害试剂和溶剂的大量使用对环境的破坏都是不可逆的[22]; 反应为非均相体系, 难于监测和分析反应进程; 增长中的肽链聚集会大大增加合成的难度[23].
此外, 在采用Fmoc策略进行肽链偶联时, Fmoc保护基的脱除与捕获也面临显著挑战. Fmoc保护基的去除是在碱的存在下通过β-消除反应实现的. 传统上, 哌啶和四氢吡咯等试剂被广泛用于脱除Fmoc保护基团, 并促进Fmoc基团衍生的二苯富烯(DBF)转化为富烯加成物. 然而, 在使用常见的Fmoc去除试剂(如哌啶和四氢吡咯)时, 未能完全捕获所有的DBF, 导致有机层中残留了较多的富烯加合物. 随后, 在用盐酸洗涤去除碱及其富烯加合物副产物时, 溶液在萃取过程中发生乳化, 导致相分离效果不理想. 推测这一现象的原因是, 用于去除富烯加合物和残留碱的盐酸在洗涤过程中引发了乳液的形成. 这可能是因为在酸性条件下, 肽的N端发生质子化, 使其表现出两亲性, 从而导致了乳化现象的产生[9,24].
为解决上述问题, 本研究开发了一种基于新型硫醇试剂3-巯基-1-丙烷磺酸钠(Mps)的DBF捕获体系. 针对Mps在乙酸乙酯(EA)中溶解度较低这一问题, 引入了二甲基亚砜(DMSO)作为共溶剂, 构建了混合溶剂体系, 实现了试剂的均匀分散. 该工艺通过与脱保护试剂1,8-二氮杂二环十一碳-7-烯(DBU)的协同作用, 生成的加成产物无需酸洗, 仅通过碱洗即可去除, 简化了后处理步骤, 同时显著降低了乳化的可能性.
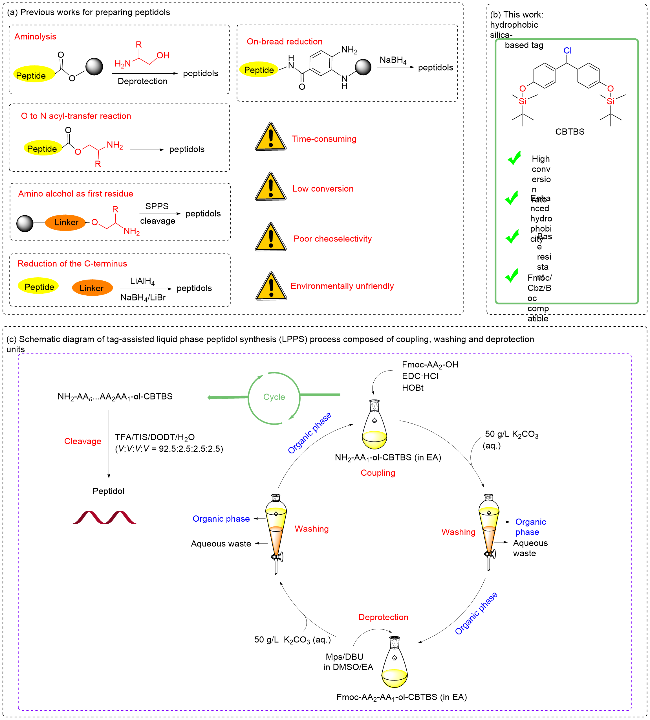
针对传统固相肽醇合成方法存在的不足, 本研究设计合成了一种新型疏水性硅基标签(CBTBS), 将其作为氨基醇类化合物C端保护基团(Scheme 1b). 该标签具有碱耐受性和疏水性, 简化了后处理步骤, 并提高了工艺效率. 此外, CBTBS标签具有易合成、易检测、低成本等特点. 基于此, 本研究开发了一种标签辅助液相肽醇合成工艺(Scheme 1c), 并合成了抗菌肽醇Gramicidin A片段(H-Trp-D-Leu-Trp-D-Leu-Trp-Gly-ol). 该工艺以Fmoc保护的氨基酸为原料, 采用3-巯基-1-丙烷磺酸钠(Mps)和1,8-二氮杂二环十一碳-7-烯(DBU)替代传统受控化学品哌啶, 实现了Fmoc基团的绿色脱除与捕获, 同时避免了萃取过程中副产物的残留和乳化问题. 标签辅助液相合成方法与传统固相工艺相比, 减少了约60%的氨基酸、偶联剂和溶剂的用量, 降低了生产成本. 同时, 以乙酸乙酯(EA)替代具有多系统毒性的N,N-二甲基甲酰胺(DMF), 进一步提升了原子经济性并降低了PMI值. 该液相合成工艺在经济性、效率、收率和灵活性等方面具有一定优势, 为绿色化学领域和工业化多肽药物的开发提供了一种解决方案.
1 结果与讨论
1.1 疏水性硅基标签的合成
疏水性硅基标签(CBTBS)以及接入第一个氨基醇的合成步骤如Scheme 2所示.
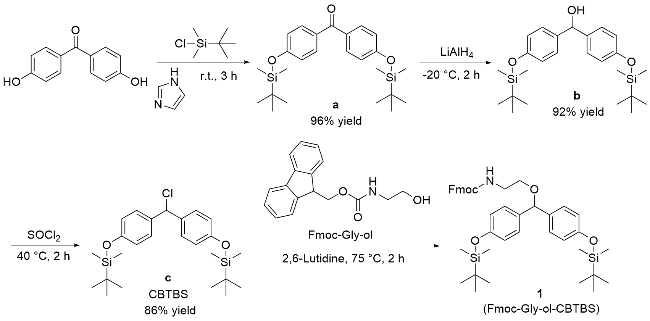
1.2 Fmoc脱保护及副产物的去除
1.2.1 哌啶、四氢吡咯与Mps捕获DBF的效果对比
为探究不同试剂对DBF的捕获效果, 本研究以Fmoc-Gly-ol-CBTBS (1) (0.71 g, 1 mmol)为反应底物, 分别在三组不同条件下进行反应: 第一组将其加入20 mL体积分数为20%的Piperidine/EA溶液; 第二组加入20 mL体积分数为20%的Pyrrolidine/EA溶液; 第三组将其与Mps (0.90 g, 5 mmol)和DBU (0.46 g, 3 mmol)共同溶解于20 mL DMSO/EA (V∶V=1∶1)的混合溶剂中. 所有反应均在30 ℃下进行20 min, 结果如表1所示.
表1 哌啶、四氢吡咯和Mps捕获DBF的效果对比Table 1 Comparison of DBF capture performance among piperidine, pyrrolidine and Mps |
| Reagent | DBF/Fulvene adducta | Washing solvent | Removal rateb/% | Yieldc/% |
|---|---|---|---|---|
| Piperidine | 6∶94 | HCl aq | 100 | 89 |
| Pyrrolidine | 16∶84 | HCl aq | 100 | 76 |
| DBU/Mps | >99 | K2CO3 aq | 100 | 98 |
a DBF/Fulvene adduct and b removal rate were determined by HPLC analysis. c Yield was determined by 1H NMR analysis. |
当以哌啶和四氢吡咯作为脱除Fmoc保护基并捕获DBF的试剂时, 均无法实现对DBF的完全捕获. 哌啶和四氢吡咯分别与DBF生成的加成产物, 都需借助体积分数为3%的盐酸来去除. 然而, 在这个过程中极易引发乳化问题. 与之相比, Mps可实现对DBF的全部捕获, 而且生成的加成产物3仅需用50 g/L的K₂CO3水溶液洗涤, 便可实现高效去除. 该过程避免了酸性条件下的质子化效应, 从根源上解决了液相乳化难题. 同时, 在此过程中, DMSO可通过水洗除去, 且所得产物不会随DMSO进入水相造成损失.
1.2.2 Fmoc脱保护反应的条件优化
以Fmoc-Gly-ol-CBTBS (1) (0.71 g, 1 mmol)为反应底物进行脱保护条件优化, 结果如表2所示. 当Mps (0.36 g, 2 mmol)与DBU (0.15 g, 1 mmol)一同溶于20 mL DMSO/EA (V∶V=2∶8)混合溶剂中, 该体系仍保持优异的脱保护效率及良好的DBF捕获效果.
表2 Fmoc脱保护反应条件优化Table 2 Optimization of Fmoc deprotection reaction conditions |
| Reagent (mmol) | DBU/mmol | DMSO/EA (volume ratio) | DBF/Fulvene adducta | Washing solvent | Removal rateb/% | Yieldc/% |
|---|---|---|---|---|---|---|
| Mps (5) | 3 | 1∶1 | >99 | K2CO3 (aq.) | 100 | 98 |
| Mps (5) | 1 | 1∶1 | >99 | K2CO3 (aq.) | 100 | 98 |
| Mps (5) | 1 | 3∶7 | >99 | K2CO3 (aq.) | 100 | 98 |
| Mps (5) | 1 | 2∶8 | >99 | K2CO3 (aq.) | 100 | 98 |
| Mps (2) | 1 | 2∶8 | >99 | K2CO3 (aq.) | 100 | 98 |
a DBF/Fulvene adduct and b removal rate were determined by HPLC analysis. c Yields were determined by 1H NMR analysis. |
综上所述, DBU/Mps体系在DMSO/EA混合溶剂中展现出显著优势. 该体系不仅能够实现对DBF的完全捕获, 而且其生成的加成产物仅通过碱洗便可彻底去除, 成功解决了传统酸洗过程中常见的乳化难题. 同时, 在这一处理过程中, DMSO可通过水洗有效除去, 且产物不会因随DMSO进入水相而造成损失.
鉴于DBU/Mps体系具备高效、简便的后处理特性, 以及良好的环境友好性, 该体系有望作为一种新型绿色脱保护工艺取代传统的哌啶体系, 在工业合成领域中得到广泛应用.
1.3 缩合试剂的选择
以NH2-Gly-ol-CBTBS (1)和Fmoc-Trp(Boc)-OH为模板反应, 合成了Fmoc-Trp(Boc)-Gly-ol-CBTBS (4)(图1). 首先在室温下, 将Fmoc-Trp(Boc)-OH (0.55 g, 1.05 mmol)与不同缩合试剂[1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDC•HCl)、2-(7-氮杂苯并三氮唑)- N,N,N',N'-四甲基脲六氟磷酸酯(HATU)、N,N'-二异丙基碳二亚胺(DIC)]在乙酸乙酯中预活化30 min, 随后分别加入NH2-Gly-ol-CBTBS (1) (0.49 g, 1 mmol)和防消旋试剂HOBt (0.15 g, 1.1 mmol)进行缩合反应, 结果见表3. HPLC检测显示反应转化率均达到100%, 考虑到后处理的便利性, 仅EDC•HCl可通过水洗直接去除, 其余缩合试剂则需经复杂的后处理步骤才能脱除. 因此, 相较于反应底物, 选择1.1 mmol的EDC•HCl作为缩合试剂为最佳方案.
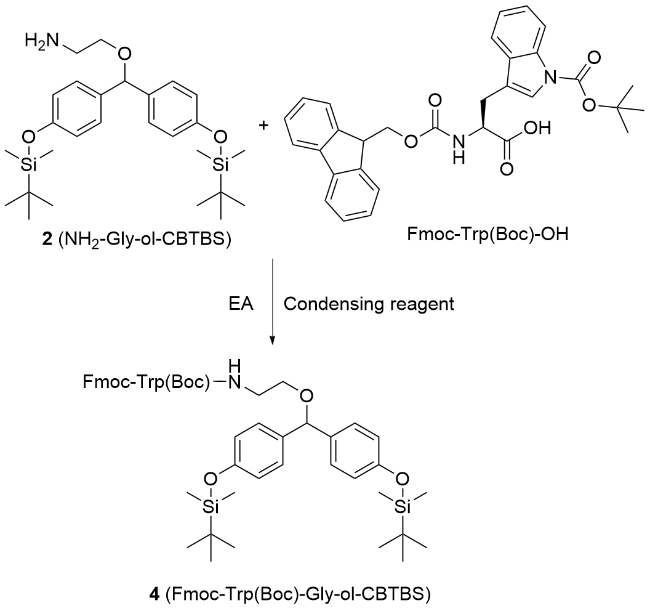
表3 缩合试剂的选择Table 3 Selection of condensing reagents |
| Entry | Coupling reagent (mmol) | Conversiona/% | Yieldb/% |
|---|---|---|---|
| 1 | HATU (1.5) | 100 | 93 |
| 2 | DIC (1.5) | 100 | 90 |
| 3 | EDC•HCl (1.5) | 100 | 98 |
| 4 | EDC•HCl (1.1) | 100 | 98 |
| 5 | EDC•HCl (1.0) | 96 | 92 |
a Conversion was determined by HPLC analysis. b Yield was determined by 1H NMR analysis. |
1.4 Gramicidin A片段的合成
1.4.1 疏水性硅基标签辅助液相合成Gramicidin A片段
Gramicidin A片段的肽序为H-Trp-D-Leu-Trp-D- Leu-Trp-Gly-ol, 使用CBTBS辅助液相合成Gramicidin A片段, 其中所需的氨基醇/酸均为市售的Fmoc保护基氨基醇/酸, 具体的操作步骤如Scheme 4所示.
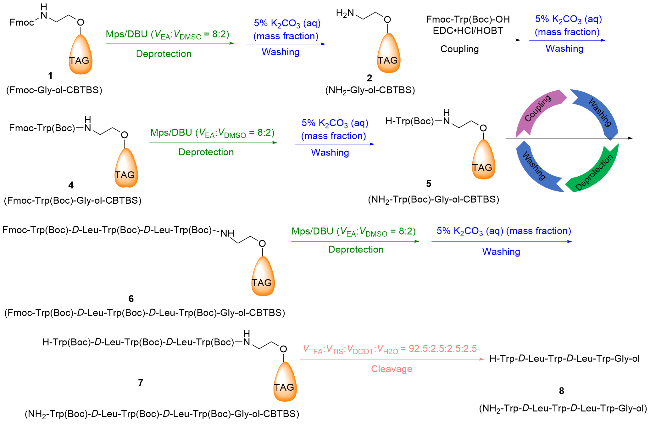
1.4.2 固相合成与标签辅助液相合成Gramicidin A片段对比
1.4.2.1 物料消耗对比
以合成1 mol的Gramicidin A片段为例, 通过对比分析固相肽醇合成(SPPS)与标签辅助液相肽醇合成(Tag-assisted LPPS)的物料消耗情况(表4), 结果表明: Tag-assisted LPPS在液-液均相反应体系中实现高效转化, 显著降低了过量氨基酸及偶联试剂的用量, 与SPPS相比, 符合绿色化学的可持续发展原则.
表4 合成Gramicidin A片段物料消耗对比Table 4 Comparison of material consumption for synthesizing Gramicidin A fragment |
| SPPS | Tag-assisted LPPS | |||
|---|---|---|---|---|
| Material | Dosage/kg | Material | Dosage/kg | |
| CTC Resin (1 mmol/g) | 1.00 | CBTBS | 0.46 | |
| Fmoc-AA-OH | 8.32 | Fmoc-AA-OH | 2.74 | |
| Coupling reagent | 5.47 | Coupling reagent | 1.09 | |
| DMF | 64.23 | EA | 2.71 | |
| Piperidine | 5.89 | DBU/Mps | 3.08 | |
此外, 值得强调的是, Tag-assisted LPPS技术采用具有生物相容性的乙酸乙酯(EA)作为反应介质, 替代了传统固相合成中具有潜在生殖毒性的N,N-二甲基甲酰胺(DMF). CBTBS在碱性条件下展现出优异的化学稳定性, 通过采用DBU/Mps体系进行脱保护反应, 有效规避了传统固相合成中Fmoc方案所需的剧毒管制化学品哌啶的使用需求. 基于上述研究结果, Tag-assisted LPPS技术在肽醇合成领域展现出显著的技术优势: 通过创新的疏水性硅基标签策略, 实现了更高的合成效率与原子经济性. 该合成体系通过采用环境友好的溶剂体系和低毒试剂组合(如EA替代DMF、DBU/Mps替代哌啶), 在保持反应高效性的同时, 显著改善了合成工艺的绿色安全性, 完全符合绿色化学的原则. 本研究为肽醇类化合物的合成提供了兼具经济性与环保性的新型解决方案.
1.4.2.2 收率与纯度对比
基于1 mol Gramicidin A片段的合成实验(表5), 系统对比了SPPS与Tag-assisted LPPS的工艺参数. 结果显示: SPPS技术需用3~5 equiv.氨基酸及偶联试剂投料. 与之形成鲜明对比的是, Tag-assisted LPPS技术通过化学计量比投料策略, 在保持高效反应的同时, 显著降低生产成本并提升原子经济性. 上述特性表明, Tag- assisted LPPS技术在满足工业级生产需求的同时, 完全契合绿色化学关于资源高效利用与工艺可持续发展的核心要求.
表5 SPPS和Tag-assisted LPPS合成Gramicidin A片段的收率与纯度对比Table 5 Comparison of yield and purity in Gramicidin A fragment synthesis: SPPS vs Tag-assisted LPPS |
| Synthetic method | Purity (HPLC)/% | Yield/% |
|---|---|---|
| SPPS | 70 | 95 |
| Tag-assisted LPPS | 65 | 80 |
1.4.2.3 PMI对比
过程质量强度(PMI)作为绿色化学评价体系的核心参数, 定义为生产过程或单元操作中总投入质量与目标产物质量的比值(Total mass input/Target product mass)[25]. 该指标通过量化物料转化效率, 有效反映化学反应工艺的可持续性水平. 一般而言, PMI值与工艺的环境友好性呈负相关, 较低的PMI值意味着更高的资源利用效率和更优的可持续发展性能, 符合现代化学工业对节能减排的技术要求.
以本文合成1 mol Gramicidin A片段为例, 将SPPS和Tag-assisted LPPS在合成阶段的PMI值进行对比(Figure 2). 结果表明, Tag-assisted LPPS的PMI值远远低于SPPS合成. 其中, 溶剂的用量是影响PMI的关键因素之一. 在固相合成中, 由于DMF具有优异的溶解能力, 成为首选溶剂与洗涤液, 然而固相合成每接入一个氨基酸都需要5~6次的洗涤, 耗费了大量的溶剂, 约占整个合成阶段PMI值的70%以上, 且DMF已被欧洲化学品管理局(ECHA)列为高度关注物质(Substances of Very High Concern, SVHC), 具有潜在致癌性, 因此寻找DMF的替代溶剂具有重要的应用价值[26]. 此外, SPPS非均相反应中使用过量的氨基酸和偶联试剂也是其PMI值远高于Tag-assisted LPPS的主要原因之一. 相比之下, Tag-assisted LPPS相比于SPPS有着更高的原子经济性和环境友好性.
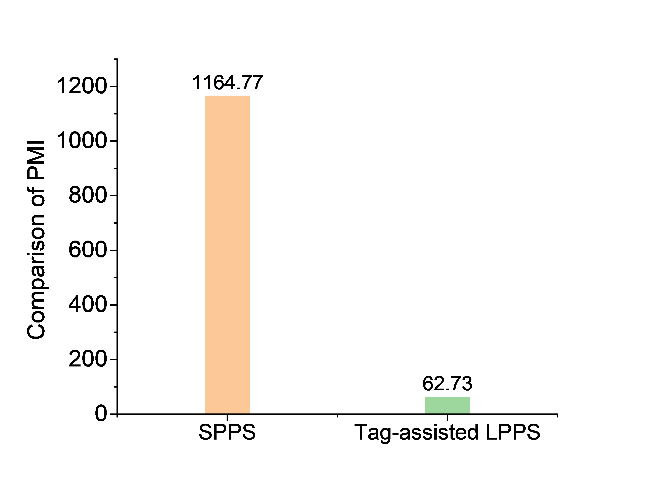
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2 结论
本研究开发了一种基于硅基疏水标签CBTBS的液相多肽合成策略, 成功实现了Gramicidin A片段的高效绿色制备. 相较于传统固相多肽合成(SPPS)技术, 本方法通过以下技术优化实现显著提升: (1)通过CBTBS标签的分子设计, 在提升肽链疏水性的同时建立了实时监测体系; (2)采用均相反应体系, 以绿色溶剂乙酸乙酯(EA)取代具有多系统毒性的N,N-二甲基甲酰胺(DMF), 并开发了基于DBU/Mps的新型脱保护体系, 替代受管制的哌啶试剂, 显著提高反应的环境友好性; 同时该工艺具备工业适配性, 工艺条件温和、放大风险小; (3)该工艺在粗肽纯度(>95%)和总收率(>80%)方面展现出优异性能, 体现了良好的原子经济性. 该策略为肽醇类药物的可持续合成提供了新途径. 此外该技术具备良好的放大潜力, 可与微反应或连续流动反应设备结合, 实现肽醇绿色高效的规模化生产.
3 实验部分
3.1 仪器与试剂
DRX500 400MHz型和ADVANCE Ⅲ 600MHz型核磁共振波谱仪(NMR), 德国Bruker公司; Agilent 1260型液相色谱-质谱联用色谱仪(LC-MS)和Agilent 1260型高效液相色谱仪(HPLC), 美国安捷伦公司; HC-2066高速离心机, 南京大卫仪器设备有限公司; N-1300型Anke Yq旋转蒸发仪、OSB-1200Anke Yq水浴锅, 上海泉杰仪器有限公司; DLK-2003快速低温冷却循环机, 宁波新芝生物科技股份有限公司.
4,4-二羟基二苯甲酮、三氟乙酸(TFA)、三异丙基硅烷(TIS)、2,2'-(1,2-乙二基双氧代)双乙硫醇(DODT), 分析纯, 上海阿拉丁试剂有限公司; 叔丁基二甲基氯硅烷、吡啶、2,6-二甲基吡啶, 四氢吡咯、氢化锂铝, 分析纯, 上海迈瑞尔化学技术有限公司; 哌啶, 分析纯, 南京晚晴化玻仪器有限公司; 氯化亚砜(SOCl2)、无水碳酸钾(K2CO3)、乙酸乙酯(EA)、甲醇(MeOH)、无水四氢呋喃(THF)、二氯甲烷(DCM)、石油醚(PE), 分析纯, 国药集团化学试剂有限公司; 3-巯基-1-丙烷磺酸钠(Mps)、1,8-二氮杂二环[5,4,0]十一烯-7 (DBU)分析纯, 上海麦克林生化科技有限公司; Fmoc氨基酸, Fmoc-Gly-ol, 分析纯, 上海吉尔生化有限公司; 1-(3-二甲基丙基)-3-乙基碳二亚胺盐酸盐(EDC•HCl)和1-羟基苯并三唑(HOBt), 分析纯, 苏州昊帆生物科技有限公司; 娃哈哈纯净水.
3.2 实验方法
3.2.1 双(4-((叔丁基二甲基硅基)氧基)苯基)甲酮(a)的合成
将4,4'-二羟基二苯甲酮(4.28 g, 20 mmol)、吡啶(6.33 g, 80 mmol)及叔丁基二甲基氯硅烷(9.04 g, 60 mmol)溶于150 mL DCM中, 反应3 h. 反应完成后, 将混合物转移至分液漏斗, 用等体积的50 g/L的K2CO3水溶液洗涤(150 mL×4), 收集有机相后减压浓缩除去溶剂, 残留物溶于200 mL MeOH中冷却析晶, 经重结晶纯化得8.49 g白色固体化合物a, 分离产率为96%. 1H NMR (400 MHz, Chloroform-d) δ: 7.65 (d, J=8.7 Hz, 4H), 6.81 (d, J=8.7 Hz, 4H), 0.87 (d, J=37.5 Hz, 18H), 0.08 (d, J=65.1 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ: 194.83, 159.60, 132.27, 131.32, 119.72, 25.71, 18.35, -4.24; HRMS-ESI calcd for C25H38NNaO3Si2 [M+Na]+ 465.1618, found 465.1644.
3.2.2 双(4-((叔丁基二甲基甲硅烷基)氧基)苯基)甲醇(b)的合成
将烘干的三口烧瓶置于杜瓦瓶中进行氮气置换, 并将8.49 g化合物a (19.20 mmol)溶解于200 mL THF中, 使用注射器注射到三口烧瓶中, 在杜瓦瓶中加入液氮, 直至温度到达零下20 ℃时, 在三口烧瓶中缓慢滴加1.46 g LiAlH4 (38.4 mmol), 反应2 h. 反应结束后, 饱和氯化铵溶液淬灭反应, 过滤出凝胶状物质后, 将滤液减压浓缩后加入150 mL EA, 用等体积的50 g/L的K2CO3水溶液洗涤(150 mL×4), 收集有机相并减压浓缩和柱层析纯化[洗脱剂为V(EA)∶V(PE)=1∶15]得到7.85 g透明油状物b, 产率92%. 1H NMR (400 MHz, Chloroform-d) δ: 7.06 (dd, J=8.5, 6.2 Hz, 4H), 6.67 (dd, J=8.5, 4.3 Hz, 4H), 5.57 (s, 1H), 0.87 (s, 18H), 0.08 (d, J=2.9 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ: 154.82, 136.92, 135.27, 128.62, 127.82, 119.94, 119.72, 79.09, 75.49, 25.74, 18.22, -4.32; HRMS-ESI calcd for C25H40- KO3Si2 [M+K]+ 483.2121, found 483.2096.
3.2.3 ((氯亚甲基)双(4,1-亚苯基))双(氧基)双(叔丁基二甲基硅烷)(c)的合成
将7.85 g化合物b (17.66 mmol)溶解于150 mL DCM中, 在0 ℃下缓慢加入3.23 g SOCl2 (27.12 mmol), 滴加完后于40 ℃下反应2 h. 反应结束后用等体积的50 g/L的K2CO3水溶液洗涤涤(150 mL×3), 之后减压浓缩和柱层析纯化[洗脱剂为V(EA)∶V(PE)=1∶10]得到7.03 g透明油状物c, 产率86%. 1H NMR (400 MHz, Chloroform-d) δ: 7.06~6.97 (m, 4H), 6.61 (d, J=8.5 Hz, 4H), 5.56 (s, 1H), 0.79 (s, 15H), 0.73 (s, 3H), -0.00 (s, 10H), -0.09 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ: 154.53, 138.74, 127.97, 119.94, 58.68, 25.80, 18.28, -4.30; HRMS-ESI calcd for C25H39O2Si2 [M-Cl]- 427.2462, found 427.2492.
3.2.4 化合物1的合成
将Fmoc-Gly-ol (0.62 g, 2.2 mmol)、2,6-二甲基吡啶(0.24 g, 2.2 mmol)和化合物c (0.92 g, 2.0 mmol)溶解于20 mL EA中, 75 ℃下反应2 h. 经HPLC检测反应完成后, 加入等体积的50 g/L的K2CO3水溶液洗涤(20 mL×3), 之后经减压浓缩和柱层析纯化[洗脱剂为V(EA)∶ V(PE)=1∶15]得到白色固体(9H-芴-9-基)甲基-2-(双(4-(叔丁基二甲基硅氧基)苯基)甲氧基)乙基氨基甲酸酯(Fmoc-Gly-ol-CBTBS, 1) 1.35 g, 1.9 mmol). 1H NMR (400 MHz, Chloroform-d) δ: 7.77 (d, J=7.5 Hz, 2H), 7.60 (d, J=7.5 Hz, 2H), 7.40 (t, J=7.4 Hz, 2H), 7.34~7.27 (m, 2H), 7.20~7.09 (m, 4H), 6.87~6.64 (m, 4H), 5.26 (s, 1H), 5.18 (s, 1H), 4.38 (d, J=7.1 Hz, 2H), 4.23 (t, J=7.1 Hz, 1H), 3.52 (t, J=5.0 Hz, 2H), 3.44 (t, J=5.2 Hz, 2H), 0.97 (s, 18H), 0.18 (s, 12H); 13C NMR (101 MHz, Chloroform-d) δ: 156.23, 154.85, 143.79, 141.10, 134.48, 127.99, 127.50, 127.47, 126.84, 124.91, 119.77, 119.66, 83.01, 67.55, 66.55, 47.04, 40.92, 25.46, 17.96, -4.61; HRMS- ESI calcd for C42H55NNaO5Si2 [M+Na]+ 732.3546, found 732.3571.
3.2.5 化合物2的合成
将Fmoc-Gly-ol-CBTBS (1, 1.35 g, 1.9 mmol)溶解在20 mL的DMSO/EA (V∶V=2∶8)混合溶剂中, 再加入3.8 mmol Mps和1.9 mmol DBU, 40 ℃下进行脱保护20 min, 经HPLC检测反应完全, 用等体积的5 g/L K2CO3水溶液洗涤(20 mL×3), 以去除体系中的加成产物、过量DBU和Mps, 以及偶联步骤中未彻底水洗去除的无保护基氨基酸. 减压浓缩有机相, 得到2-(双(4-(叔丁基二甲基硅氧基)苯基)甲氧基)乙胺(NH2-Gly-ol-CBTBS, 2) 0.91 g, 1.86 mmol. 1H NMR (400 MHz, Chloroform-d) δ: 7.77 (d, J=7.5 Hz, 2H), 7.60 (d, J=7.5 Hz, 2H), 7.40 (t, J=7.4 Hz, 2H), 7.34~7.27 (m, 2H), 7.20~7.09 (m, 4H), 6.87~6.64 (m, 4H), 5.26 (s, 1H), 5.18 (s, 1H), 4.38 (d, J=7.1 Hz, 2H), 4.23 (t, J=7.1 Hz, 1H), 3.52 (t, J=5.0 Hz, 2H), 3.44 (t, J=5.2 Hz, 2H), 0.97 (s, 18H), 0.18 (s, 12H); 13C NMR (101 MHz, Chloroform-d) δ: 156.23, 154.85, 143.79, 141.10, 134.48, 127.99, 127.50, 127.47, 126.84, 124.91, 119.77, 119.66, 83.01, 67.55, 66.55, 47.04, 40.92, 25.46, 17.96, -4.61; HRMS-ESI calcd for C27H45- NNaO3Si2 [M+Na]+ 510.2878, found 510.2848.
3.2.6 DBF和化合物3的表征
9-亚甲基-9H-芴(DBF): 1H NMR (400 MHz, Chloroform-d) δ: 7.69 (dd, J=16.8, 7.5 Hz, 4H), 7.32 (dtd, J=28.6, 7.5, 1.2 Hz, 4H), 6.05 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ: 143.39, 140.20, 138.08, 128.79, 127.11, 121.07, 119.81, 107.86; HRMS-ESI calcd for C14H11 [M+H]+ 179.0159, found 179.0186.
3-(((9H-芴-9-基)甲基)硫基)丙烷-1-磺酸钠(3): 1H NMR (400 MHz, Chloroform-d) δ: 7.61 (dd, J=30.6, 7.4 Hz, 4H), 7.26 (dt, J=28.1, 7.4 Hz, 4H), 3.99 (t, J=6.2 Hz, 1H), 3.02 (dd, J=48.9, 7.0 Hz, 4H), 2.49 (t, J=6.9 Hz, 2H), 2.17~1.98 (m, 2H); 13C NMR (101 MHz, Chloroform-d) δ: 145.96, 141.12, 127.71, 127.17, 124.90, 120.01, 46.90, 36.00, 31.28, 23.76; HRMS-ESI calcd for C17H18- NaO3S2 [M+H]+ 357.0134, found 357.0104.
3.2.7 化合物4和5的合成
活化氨基酸: 称取相对于化合物2 (0.91 g, 1.86 mmol)为1.05 equiv.的Fmoc-Trp(Boc)-OH (1.03 g, 1.95 mmol), 1.1 equiv.的偶联试剂EDC•HCl (0.39 g, 2.05 mmol)和HOBt (0.28 g, 2.05 mmol)溶解在20 mL EA中, 在室温下预活化30 min, 可以活化氨基酸羧基以便进行偶联反应. Gramicidin A片段序列中所需要的Fmoc-D- Leu-OH、Fmoc-Trp(Boc)-OH、Fmoc-D-Leu-OH和Fmoc- Trp(Boc)-OH使用上述方法进行活化.
将NH2-Gly-ol-CBTBS (2)溶解于Fmoc-Trp(Boc)- OH的活化液中, 40 ℃下搅拌40 min, 经HPLC检测反应完全后, 用等体积的50 g/L的K2CO3水溶液洗涤(20 mL×3), 去除体系中过量氨基酸及偶联试剂残留后, 获得目标产物Fmoc-Trp(Boc)-Gly-ol-CBTBS (4, 1.81 g, 1.82 mmol), 之后再溶解于20 mL的DMSO/EA (V∶V=2∶8)混合溶剂中, 往其加入Mps (0.65 g, 3.62 mmol)和DBU (0.28 g, 1.81 mmol), 脱保护得到NH2-Trp(Boc)- Gly-ol-CBTBS (5, 1.38 g, 1.79 mmol).
(S)-3-(2-(((9H-芴-9-基)甲氧基)羰基)氨基)-3-((2-(双(4-(叔丁基二甲基硅氧基)苯基)甲氧基)乙基)氨基)-3-氧代丙基)-1H-吲哚1羧酸叔丁酯(4): 1H NMR (400 MHz, Chloroform-d) δ: 8.17 (s, 1H), 7.78 (d, J=7.6 Hz, 2H), 7.66~7.50 (m, 3H), 7.47~7.38 (m, 2H), 7.38~7.30 (m, 3H), 7.26 (d, J=7.4 Hz, 1H), 7.06~6.93 (m, 4H), 6.74 (dd, J=8.3, 1.5 Hz, 4H), 6.02 (d, J=6.0 Hz, 1H), 5.58 (d, J=7.9 Hz, 1H), 5.02 (s, 1H), 4.50~4.41 (m, 2H), 4.39~4.31 (m, 1H), 4.15 (q, J=7.1 Hz, 1H), 3.37 (d, J=5.2 Hz, 3H), 3.28 (d, J=19.3 Hz, 1H), 3.17 (d, J=7.7 Hz, 2H), 2.07 (s, 1H), 1.64 (s, 7H), 1.34~1.25 (m, 2H), 0.98 (d, J=2.9 Hz, 18H), 0.18 (d, J=2.6 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ: 170.76, 155.99, 155.11, 155.06, 149.60, 143.83, 143.80, 141.37, 135.55, 134.59, 134.42, 130.34, 128.25, 128.17, 127.84, 127.21, 125.19, 124.85, 124.43, 122.94, 120.10, 119.93, 119.88, 119.17, 115.51, 83.84, 83.16, 67.28, 67.20, 55.44, 47.19, 28.79, 28.28, 25.75, 18.24, -4.32; HRMS-ESI calcd for C58H73N3Na- O8Si2 [M+Na]+ 1018.4889, found 1018.4860.
(S)-3-(2-氨基-3-((2-(双(4-(叔丁基二甲基硅氧基) 苯基)甲氧基)乙基)氨基)-3-氧代丙基)-1H-吲哚-1-羧酸叔丁酯(5): 1H NMR (400 MHz, Chloroform-d) δ: 8.23~8.08 (m, 1H), 7.68 (dd, J=18.5, 6.3 Hz, 2H), 7.57~7.46 (m, 1H), 7.40~7.31 (m, 1H), 7.26 (d, J=6.9 Hz, 1H), 7.15 (dd, J=8.4, 6.7 Hz, 4H), 6.79 (d, J=8.3 Hz, 4H), 5.21 (s, 1H), 4.15 (q, J=7.1 Hz, 1H), 3.73 (dd, J=9.5, 3.9 Hz, 1H), 3.61~3.34 (m, 5H), 2.83 (dd, J=14.5, 9.5 Hz, 1H), 2.07 (s, 1H), 1.69 (s, 9H), 0.99 (s, 18H), 0.20 (s, 12H). 13C NMR (101 MHz, Chloroform-d) δ: 174.42, 155.11, 134.88, 134.81, 131.04, 130.45, 128.96, 128.34, 124.76, 124.21, 122.77, 119.89, 119.41, 116.97, 115.44, 83.13, 67.72, 65.68, 60.51, 55.05, 39.11, 30.84, 28.33, 25.76, 18.27, -4.30; HRMS-ESI calcd for C38H54N3O4Si2 [M-Boc+H]+ 701.4992, found 701.4963.
3.2.8 化合物6~8的合成
肽链延伸阶段采用循环合成策略, 依次执行上述偶联与脱保护操作, 最终得到带有CBTBS标签的全保护Gramicidin A片段Fmoc-Trp(Boc)-D-Leu-Trp (Boc)-D- Leu-Trp(Boc)-Gly-ol-CBTBS (6, 3.00 g, 1.66 mmol). 之后将化合物6通过脱保护得到NH2-Trp(Boc)-D-Leu-Trp- (Boc)-D-Leu-Trp(Boc)-Gly-ol-CBTBS (7, 2.59 g, 1.65 mmol). 在30 ℃下, 采用体积比为92.5∶2.5∶2.5∶2.5的TFA-TIS-DODT-H₂O四元混合体系, 对CBTBS保护基及侧链保护基进行2 h的裂解反应, 随后通过甲基叔丁基醚沉降处理, 经离心分离和氮气吹干, 最终获得白色固体Gramicidin A片段粗肽产品NH2-Trp-D-Leu-Trp- D-Leu-Trp-Gly-ol (8) 1.35 g, 产率80%, 纯度95%.
Fmoc-Trp(Boc)-D-Leu-Trp(Boc)-D-Leu-Trp(Boc)-Gly-ol-CBTBS (6): 1H NMR (400 MHz, Chloroform-d) δ: 7.95~7.80 (m, 4H), 7.48 (q, J=7.8 Hz, 6H), 7.30 (d, J=30.3 Hz, 14H), 6.97 (q, J=6.9 Hz, 16H), 6.57~6.53 (m, 4H), 5.09 (s, 1H), 5.06 (s, 1H), 4.34 (s, 2H), 3.97 (s, 4H), 3.68 (s, 2H), 3.54 (d, J=17.0 Hz, 2H), 3.32 (d, J=11.3 Hz, 6H), 1.41 (d, J=21.7 Hz, 32H), 0.76 (d, J=2.6 Hz, 18H), 0.68~0.56 (m, 12H), -0.04 (d, J=3.8 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ: 173.99, 173.44, 172.27, 171.50, 154.56, 141.01, 136.66, 135.03, 130.60, 128.34, 128.05, 127.55, 126.85, 126.77, 124.98, 124.32, 122.70, 122.32, 119.68, 119.44, 117.27, 115.33, 115.21, 114.84, 83.06, 53.24, 46.64, 42.95, 27.98, 26.62, 25.46, 24.78, 20.92, 17.95, -4.61, -4.63; HRMS-ESI calcd for C102H131N9KO16Si2 [M+K+H]2+ 917.4478, found 917.4453.
NH2-Trp(Boc)-D-Leu-Trp(Boc)-D-Leu-Trp(Boc)-Gly-ol-CBTBS (7): 1H NMR (400 MHz, Chloroform-d) δ: 7.90 (d, J=48.5 Hz, 3H), 7.45 (d, J=7.6 Hz, 1H), 7.38~6.90 (m, 20H), 6.65~6.52 (m, 5H), 5.11 (s, 1H), 4.56 (s, 1H), 4.20 (s, 1H), 3.97 (s, 1H), 3.72 (s, 1H), 3.43~3.27 (m, 5H), 3.22 (s, 1H), 3.16 (s, 1H), 2.95 (d, J=11.8 Hz, 2H), 2.72 (d, J=14.4 Hz, 2H), 2.41 (s, 1H), 2.12 (s, 1H), 1.52~1.31 (m, 31H), 0.79 (s, 18H), 0.68~0.50 (m, 12H), -0.00 (s, 12H); 13C NMR (101 MHz, Chloroform-d) δ: 172.74, 155.20, 150.16, 135.72, 128.88, 128.78, 125.22, 124.64, 123.31, 122.95, 120.19, 120.10, 119.43, 115.86, 115.49, 84.81, 83.58, 83.31, 67.71, 54.04, 40.28, 30.12, 28.63, 26.10, 23.68, 23.19, 22.11, 21.74, 18.58, -3.96; HRMS-ESI calcd for C87H124N9O14Si2 [M+3H]3+ 525.2189, found 525.2217.
(S)-2-((S)-2-氨基-3-(1H-吲哚-3-基)丙酰胺基)-N- ((S)-1-(((S)-1-(((S)-1-((2-羟乙基)氨基)-3-(1H-吲哚-3-基)- 1-氧代丙-2-基)氨基)-4-甲基-1-氧代戊-2-基)氨基)- 3-(1H-吲哚-3-基)-1-氧代丙-2-基)-4-甲基戊酰胺(8): 1H NMR (400 MHz, DMSO-d6) δ: 10.92 (s, 1H), 10.79 (d, J=18.5 Hz, 2H), 8.45 (s, 1H), 8.19 (s, 1H), 8.09 (d, J=8.0 Hz, 1H), 7.99 (dd, J=26.2, 7.6 Hz, 1H), 7.89 (q, J=8.5, 7.1 Hz, 1H), 7.63 (t, J=8.8 Hz, 2H), 7.55 (d, J=8.1 Hz, 1H), 7.33 (dd, J=16.3, 8.1 Hz, 3H), 7.16 (d, J=7.1 Hz, 2H), 7.10 (s, 1H), 7.04 (t, J=7.7 Hz, 3H), 6.99~6.89 (m, 3H), 4.63 (dt, J=22.4, 6.4 Hz, 2H), 4.50 (q, J=6.9 Hz, 1H), 4.44~4.35 (m, 1H), 4.30 (h, J=8.2, 6.6 Hz, 2H), 3.78 (s, 1H), 2.96 (dd, J=14.7, 8.3 Hz, 2H), 2.86 (dt, J=14.6, 6.1 Hz, 1H), 1.11 (s, 6H); 13C NMR (101 MHz, DMSO-d6) δ: 171.90, 171.79, 171.40, 171.35, 168.49, 158.97, 158.62, 151.70, 136.59, 136.22, 135.13, 130.58, 127.63, 127.22, 125.32, 124.43, 123.85, 123.63, 121.32, 120.99, 120.98, 118.87, 118.57, 118.38, 117.65, 114.75, 111.62, 111.44, 110.28, 109.95, 107.03, 72.25, 53.33, 52.54, 51.44, 48.86, 41.45, 41.08, 40.15, 31.42, 29.63, 27.46, 26.95, 24.18, 23.33, 23.31, 21.81; HRMS-ESI calcd for C47H60N9O6 [M+H]+ 846.3867, found 846.3838.
辅助材料(Supporting Information) 化合物a~c、1~8和DBF的核磁与质谱图谱、不同试剂捕获DBF的液相图以及通过标签辅助液相与固相合成Gramicidin A片段的物料消耗表. 这些材料可以免费从本刊网站(http:// sioc-journal.cn/)上下载.
(Cheng, F.)