


蛋白质和多肽作为生命活动的核心功能分子, 在生命系统中发挥着不可替代的调控作用. 它们通过调控细胞信号传导、免疫应答和酶促反应等关键生物过程, 实现对生命活动的多维度调控[1]. 随着蛋白质结构与功能关系研究的不断深入, 特别是结构生物学与单分子技术的革命性进展, 其精确的结构调控机制已成为解析生命本质的关键科学问题, 同时也为开发高靶向性、高安全性的新型生物大分子药物提供了理论基础[2]. 在此背景下, 蛋白质/多肽精准合成技术作为核心驱动力, 正推动相关研究领域的快速发展. 该技术能够构建自然界不存在的或难以获取的、具有特定功能优化(如增强稳定性、提高活性、引入新型功能基团)的工程化蛋白质或多肽药物, 从而为重大疾病治疗提供创新性分子工具.
近年来, 单一位点化学修饰技术与自动化合成平台的协同发展, 显著推动了蛋白质和多肽的精准合成进程. 这些技术突破使得在分子水平上实现对蛋白质/多肽结构的精确调控成为可能, 从而深化了对构效关系的理解. 在蛋白质/多肽精准修饰领域, 通过发展基于位点特性的直接修饰、配体导向修饰及关键点定向修饰等策略, 已实现了对复杂生物大分子的单一位点精确修饰[3]. 这些技术的发展为开发高均一性的抗体药物偶联物(ADCs)等新一代生物制剂奠定了重要基础, 显著提高了治疗药物的靶向特异性和安全性. 在单一位点化学修饰技术持续提升分子结构精确调控能力的同时, 针对多位点协同修饰、大规模蛋白质库构建等更复杂、更高通量的应用需求, 整合了固相合成、微流控技术、高通量自动化平台及人工智能辅助序列设计等先进技术的新型自动化合成系统, 展现出显著的协同效应[4]. 该技术不仅能够实现含特定非天然氨基酸或其衍生物的目标蛋白质或多肽的按需精准合成, 更能显著提高复杂生物分子的合成精确度和规模化制备效率, 从而将蛋白质/多肽精准合成技术推向新的发展阶段.
本文系统评述了近五年来单一位点化学修饰与自动化合成平台的协同创新对蛋白质及多肽精准合成领域的推动作用. 这些技术突破不仅克服了传统方法在修饰位点控制和合成效率方面的局限性, 更通过“精准化学”研究范式实现了蛋白质结构的可控合成, 为开发具有增强稳定性、提高靶向性及创新功能活性的生物治疗分子提供了关键技术支撑.
1 蛋白质/多肽单一位点选择性化学修饰
蛋白质/多肽的精准化单一位点选择性修饰与标记是化学生物学领域实现分子功能定向改造的关键, 其核心目标是在复杂生物分子环境中实现对特定一个氨基酸残基或位点的高效、精准修饰. 早期的化学修饰方法往往缺乏位点特异性, 难以满足基础研究和生物医药应用的需求. 近年来, 通过理性设计反应试剂或利用蛋白质局部微环境特性, 研究者已开发出多种位点特异性修饰策略[3]. 本节将系统总结近五年来蛋白质/多肽单一位点选择性修饰的研究进展, 包括基于位点特性的直接修饰技术、配体导向策略及关键点定向方法.
1.1 基于位点特性的直接修饰技术
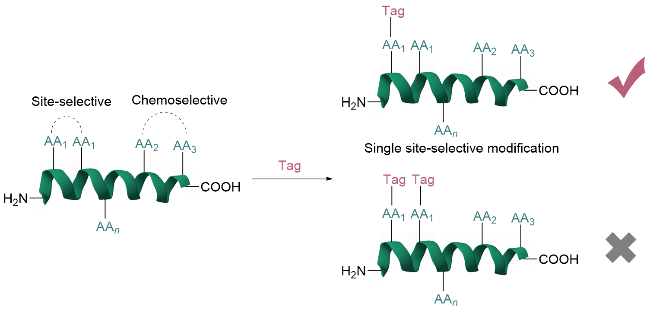
对蛋白质/多肽序列中的高活性氨基酸进行直接修饰是一种简便高效的策略, 但该方法常因序列中存在多个同种氨基酸残基而导致非特异性修饰(图1). 基于特殊位点独特结构或化学环境的选择性修饰技术解决了这一问题. 该技术通过理性设计反应试剂或优化反应条件, 使修饰反应优先发生在特定位点, 从而实现对蛋白质/多肽的单一位点精准标记. 例如, Bernardes和Jiménez-Osés等[5]通过计算机辅助设计磺酰丙烯酸酯试剂, 实现了对天然蛋白质中单个赖氨酸残基的高效、区域选择性修饰. 该方法基于赖氨酸局部微环境赋予的独特反应性差异, 通过试剂中磺酰基与目标赖氨酸ε-氨基形成的氢键网络稳定椅式过渡态, 显著降低了反应能垒, 并选择性地靶向pKa最低的赖氨酸残基[如重组人血清白蛋白(HSA)中的K573和溶菌酶中的K33], 实现动力学控制的单个赖氨酸残基修饰. 尽管该策略在特定蛋白中效果显著, 但其普适性仍受蛋白质特定结构的限制. 相较而言, 针对多肽和蛋白质末端的位点选择性修饰展现出更显著的优势. 其原因在于, 单链蛋白质仅含有一个N端或C端, 位点特异性明确; 该位点通常暴露于溶剂环境中; 特别是N端α-氨基与赖氨酸侧链ε-氨基的pKa差异显著. 这些特性使末端选择性修饰技术成为蛋白质修饰改造中非常具有发展前景的修饰策略, 尤其在需要保持蛋白质整体结构的应用中具有独特价值[6].
1.1.1 N端氨基单一位点修饰
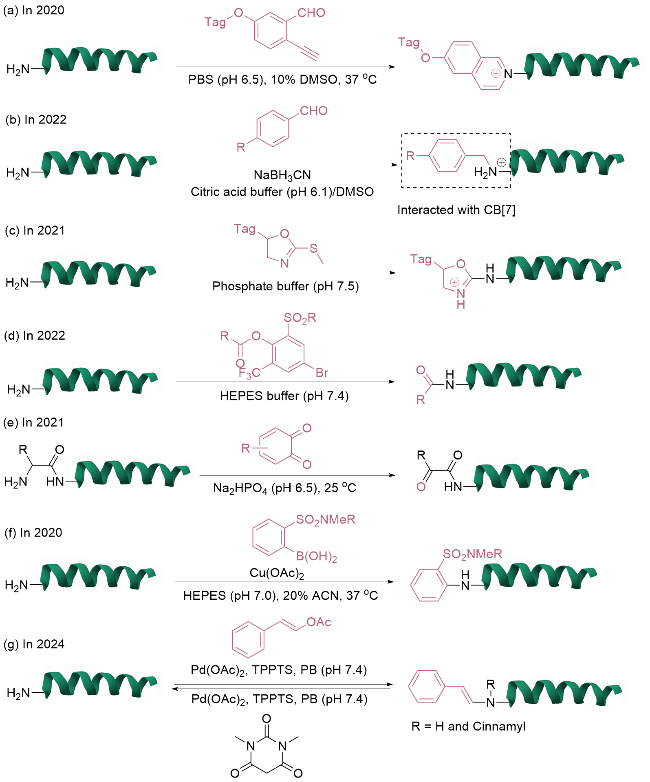
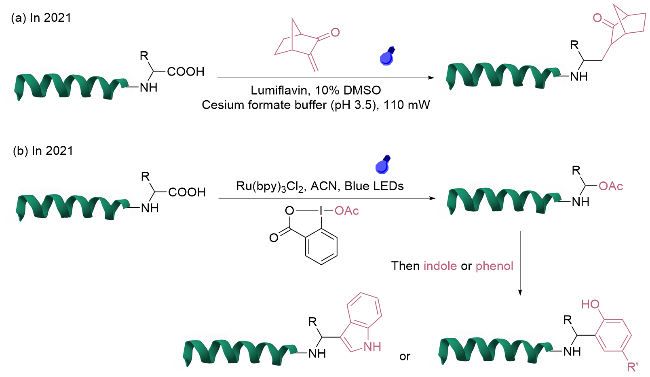
对于N端氨基的单一位点修饰, 关键在于区分对赖氨酸氨基的反应活性, 传统缩合反应往往导致多位点修饰产物的生成. 开发新型试剂或方法可以使修饰在动力学或热力学上优先作用于N端氨基. 利用醛基与氨基的高效缩合反应特性, Wong团队[7]在2020年研究了2-乙炔基苯甲醛(2-EBA)在多肽和蛋白质N端选择性修饰中的应用及其机制, 通过系统筛选反应条件和2-EBA的结构与活性关系, 发现电子效应和pH调控对N端修饰的选择性具有重要影响(图2a). 在弱酸性条件(pH=6.5)下, 带有供电子基团的2-EBA表现出优异的N端选择性(>99:1), 而碱性条件(pH=9.0)和吸电子基团使反应更倾向于赖氨酸ε-氨基的修饰. 进一步的机理研究表明, 2-EBA与N端α-氨基通过亚胺中间体形成稳定的异喹啉盐结构, 该修饰产物在氧化还原环境中表现出良好的稳定性. 该方法能够兼容除N端脯氨酸外的其他N端天然氨基酸残基, 并且对溶菌酶、核糖核酸酶A以及治疗性重组蛋白等多种生物大分子的修饰均展现出良好的适用性.
1.1.2 N端主链酰胺键单一位点修饰
2023年, Spicer等[17]通过比较2-吡啶甲醛、三唑甲醛、2-乙炔基苯甲醛等7种代表性试剂在核糖核酸酶A、肌红蛋白和梭菌蛋白酶轻链等模型蛋白上的修饰效率、选择性和稳定性, 揭示了这7种试剂在当前N端修饰策略的优势与局限性. 研究表明, 蛋白质局部微环境(如空间位阻、pKa)会显著影响修饰效果, 目前尚无普适性N端修饰策略, 需根据目标蛋白特性进行试剂筛选.2025年, 该团队[18]系统研究了12种吡啶甲醛衍生物在蛋白质N端选择性修饰中的反应性, 重点分析了半胺缩醛、亚胺和咪唑烷酮生成三个关键步骤的动力学与热力学影响因素, 强调需针对特定蛋白筛选最优试剂, 其中3-甲氧基-2-吡啶甲醛因其普适性成为最具潜力的候选分子, 为设计新一代N端标记试剂提供了重要指导.
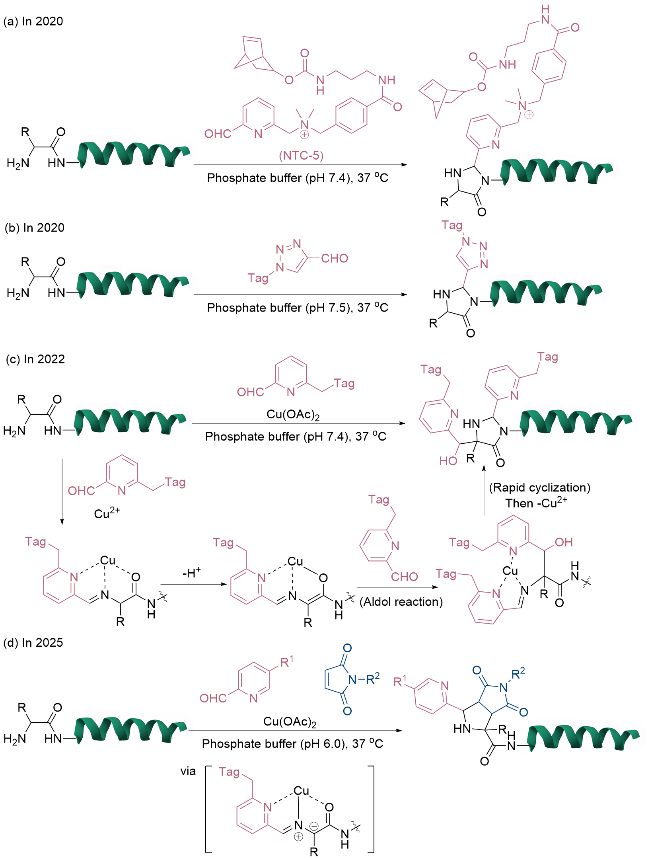
2022年, Hanaya等[19]开发了一种基于铜(II)介导的Aldol反应的N端修饰方法. 2-吡啶甲醛衍生物与蛋白质N端氨基缩合形成亚胺, 并在铜(II)离子配位作用下形成稳定的席夫碱-铜(II)三元复合物. 该复合物有效活化N端氨基酸的α-质子生成亲核中间体, 进而与醛类化合物发生Aldol反应形成稳定的碳-碳键. 随后, Thorpe-Ingold效应促进分子内酰胺氮对亚胺碳的亲核进攻, 经环化反应形成稳定的咪唑烷酮结构, 最终通过乙二胺四乙酸(EDTA)螯合实现铜离子的脱除(图3c). 基于这一机制, 他们[20]在2025年开发了基于铜(II)介导[3+2]环加成反应的N端双功能化策略(图3d). 研究发现, 2-吡啶甲醛衍生物、N端氨基酸与铜(II)离子可协同形成具有1,3-偶极子特性的活性中间体, 该物种能与马来酰亚胺类化合物发生区域选择性的环加成反应, 实现对N端不同氨基酸的高效修饰. 该方法成功应用于构建三元蛋白质复合物和双功能化抗体药物偶联物, 修饰后的抗体不仅保持了人表皮生长因子受体2 (HER-2)结合活性, 还表现出显著的抗肿瘤效果和体内成像能力.
1.1.3 N端脯氨酸单一位点修饰

1.1.4 N端甘氨酸单一位点修饰
1.1.5 N端半胱氨酸单一位点修饰

1.1.6 C端单一位点修饰
1.2 配体导向的单一位点选择性修饰技术
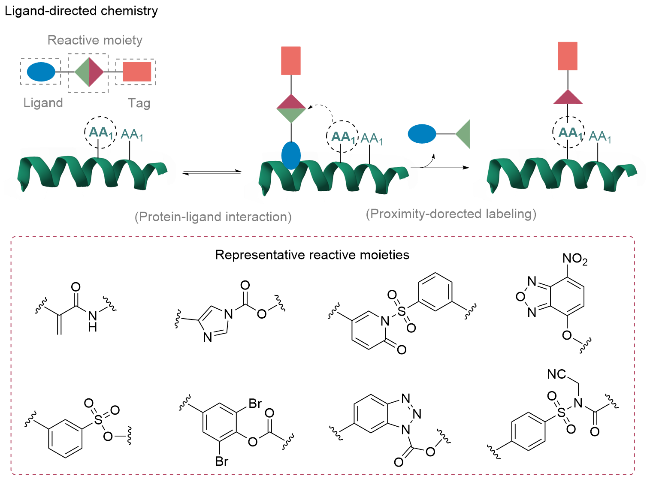
在天然蛋白质单一位点标记领域, 当直接修饰无法满足选择性需求时, 基于蛋白质与配体识别邻近效应的配体导向修饰策略是一种重要技术手段[35](图8). 该策略采用含与蛋白质结合的配体、反应性官能团以及修饰/标记的分子模块三个关键组分的多功能试剂, 通过配体模块特异性识别并结合目标蛋白、亲电反应性基团与邻近亲核氨基酸残基发生选择性反应、配体与蛋白复合物解离三个连续步骤, 从而实现对目标蛋白的特异性单一位点修饰. 对于蛋白质中的不同亲核氨基酸残基的选择性修饰位点, 发展了包括甲苯磺酰基、酰基咪唑、苯甲酸二溴苯酯、N-磺酰基吡啶酮、苯并三唑、O-硝基苯并恶二唑、N-酰基-N-烷基磺酰胺等反应性官能团或方法[3h,36](图8). 这些反应基团通过特异性识别不同活性位点, 实现了蛋白质多样化精准修饰.
1.2.1 配体导向赖氨酸单一位点修饰
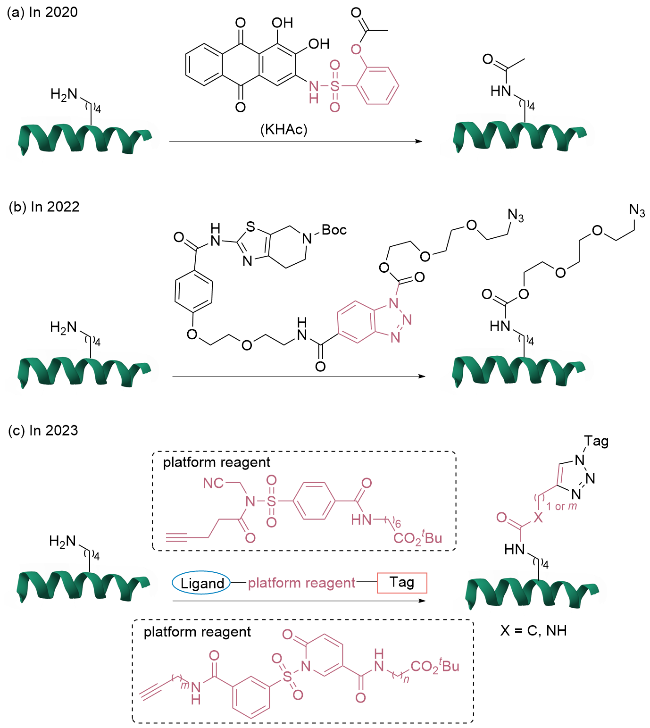
1.2.2 基于共价配体定向释放策略的半胱氨酸单一位点修饰
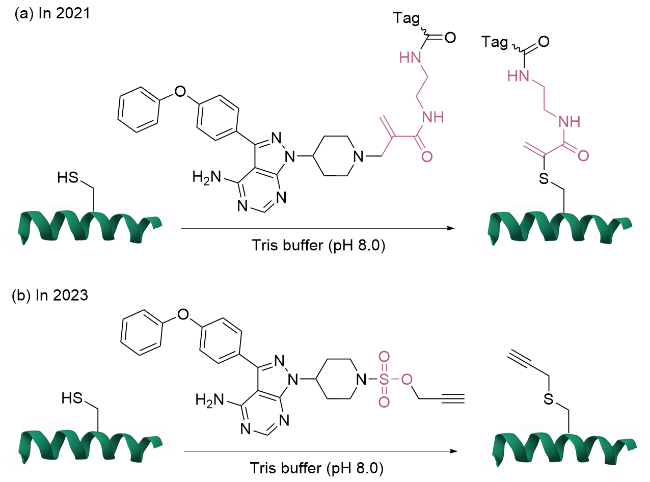
1.2.3 基于亲和力引导催化剂策略的单一位点修饰

1.3 关键点定向修饰技术

1.3.1 赖氨酸导向赖氨酸单一位点修饰
利用该技术完成对天然蛋白质中组氨酸残基[46]的高效位点选择性修饰后, Rai团队[47]在2020年将该技术通过双功能试剂(FK1-spacer-FK2)拓展至天然蛋白质中高频率赖氨酸残基的单一位点标记(图13a). 在该方法中, 2-羟基苯甲醛基团(FK1)通过可逆的亚胺结构与溶剂可及的赖氨酸残基结合, 随后相邻的赖氨酸残基与酰化亲电试剂(FK2)通过分子内反应实现不可逆的位点选择性修饰, 克服了传统赖氨酸修饰中的异质性问题. 该技术不仅适用于核糖核酸酶A、胰岛素、抑肽酶、泛素、α-乳清蛋白、细胞色素c和肌红蛋白等蛋白质, 还能在抗体-药物偶联物(ADCs)中实现高效应用, 例如将抗癌药物DM1与曲妥珠单抗偶联, 所得偶联药物在HER-2阳性乳腺癌细胞中表现出高度特异性的抗增殖活性. 2024年, 该团队[48]进一步优化该技术, 并将其应用于胰蛋白酶和α-糜蛋白酶的定点修饰, 不仅有效避免了蛋白酶的自降解问题, 还通过醛基标签实现了酶分子的有序固定化. 与传统的随机固定化方法相比, 这种定点固定化策略显著提升了蛋白酶的消化效率, 并在多次循环使用后仍能保持较高的催化活性.
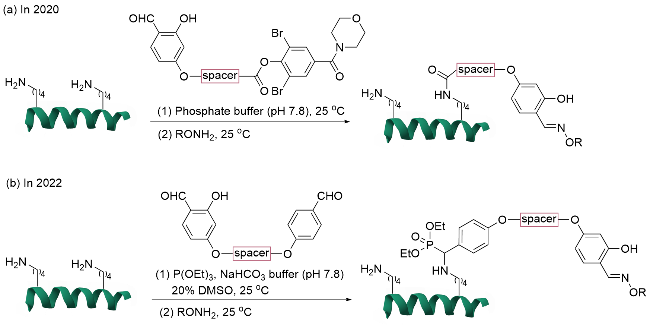
基于这一策略, 他们[49]在2022年重新设计了双醛基团结构的双功能试剂, 利用化学正交的亚胺反应性差异, 实现了对特定赖氨酸残基的位点选择性标记(图13b). 其中, 2-羟基苯甲醛基团(FK1)与特定赖氨酸形成的亚胺保持化学惰性, 而苯甲醛基团(FK2)与邻近赖氨酸生成活性亚胺, 与外部亲核试剂(三乙氧基磷)选择性发生不可逆反应, 从而完成单一位点修饰. 研究通过理论计算和实验验证, 证实了2-羟基苯甲醛基团形成的亚胺因π共轭稳定化而惰性, 而苯甲醛基团形成的亚胺表现出与外部亲核试剂更高的反应性, 从而实现了化学正交性调控. 此外, 该技术展现出优异的模块化能力, 通过调整双功能试剂的空间构象, 可靶向不同位置的赖氨酸(如细胞色素c中的K60和K27), 且不干扰蛋白质的天然结构和酶活性, 展示出其通过微环境调控和空间邻近效应实现精准修饰的能力.
1.3.2 半胱氨酸导向赖氨酸单一位点修饰
2022年, Rai团队[50]开发了一种半胱氨酸锚定导向的赖氨酸单一位点精准修饰策略(图14a). 该方法利用硝基烯烃(FC1)与蛋白质中的半胱氨酸残基发生特异性迈克尔加成反应形成共价加合物, 随后通过分子内反应使邻近的赖氨酸残基与酰化亲电试剂(FK2)实现高效且不可逆的位点选择性修饰. 随后, 硝基烯烃在温和条件下转化为可进一步正交修饰的醛基, 为后续正交功能化修饰提供了便利的化学手柄. 该研究通过分子动力学模拟研究揭示了试剂构象的适应性刚性对位点选择性的调控机制, 并被应用于多种蛋白质(如β-乳球蛋白、血清白蛋白)和抗体(如曲妥珠单抗)的修饰以及抗体-药物偶联物的制备. 同时, 该策略可以实现对复杂蛋白质混合物中单一蛋白质的单一赖氨酸残基的高效标记, 在含有57个赖氨酸残基的人血清白蛋白与其他蛋白质的混合物中, 可选择性标记人血清白蛋白中的单一赖氨酸残基.
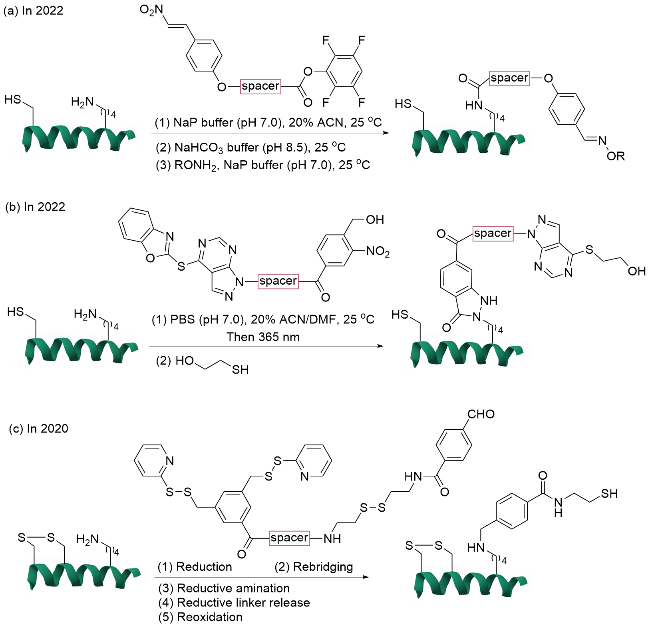
1.3.3 其他氨基酸关键点定向修饰
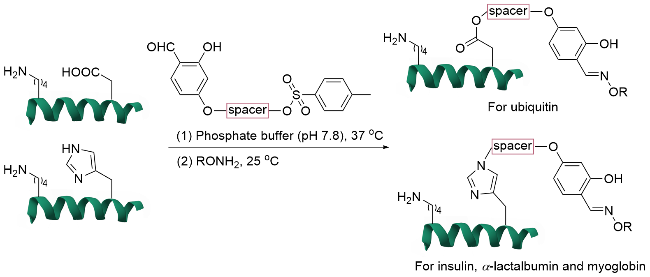
利用LDM技术, 除了对赖氨酸以及组氨酸的单一位点修饰外, Rai研究团队[53]在2021年还实现了对天冬氨酸或组氨酸的高效位点选择性修饰(图15). 该研究设计了一种独特的双功能试剂(FK1-spacer-FD2), 其结构整合了2-羟基苯甲醛基团和非选择性亲电基团(芳基磺酸酯), 有效克服了传统修饰方法反应选择性不好的问题. 研究通过系统筛选双功能试剂结构和反应条件, 优化了双功能分子的效率, 实现了泛素中天冬氨酸以及胰岛素、α-乳清蛋白和肌红蛋白中组氨酸的单一位点修饰. 并通过后期功能化成功引入19F NMR探针、生物素或荧光基团等重要功能分子, 为低丰度氨基酸的精准标记提供了实用性工具.
蛋白质/多肽的单一位点选择性化学修饰技术在蛋白质或多肽的特定位置引入结构多样、功能各异的分子, 为精准调控生物大分子功能提供了强大工具. 然而, 这些精准修饰的实现高度依赖于对反应位点的精确控制与高效的特异性反应体系, 使其难以应对多位点协同修饰或大规模蛋白质库构建等复杂需求. 而随着固相合成、液相分段合成等技术的迭代升级, 特别是自动化合成仪器的进步, 使得在合成过程中直接引入非天然氨基酸或特定修饰基团成为可能. 这种合成与修饰的融合策略, 不仅突破了传统“先合成后修饰”流程中存在的位点冲突问题, 更通过计算机控制的精确加料系统和实时监测模块, 实现了从氨基酸序列构建到位点特异性修饰的一体化精准操作. 这种技术层面的深度整合, 标志着蛋白质/多肽精准合成正从传统的分步化学操作向集成化、智能化的平台转变.
2 蛋白质/多肽自动化精准合成
2.1 自动化精准合成技术
近年来, 流动化学与多肽固相合成(SPPS)的融合创新显著推动了多肽与蛋白质合成技术的发展, 在合成效率、精准度和自动化水平等方面取得系列突破. 2014年, Pentelute团队[4b]基于流动合成多肽路线, 开发了自动化控制的流动化学合成平台(图16a), 通过高效热交换器与新反应器实现试剂的连续预热与高浓度输送, 同时通过紫外检测实时监控9-芴基甲氧基羰基(Fmoc)脱保护与偶联进程, 将脱保护时间优化至10 s, 偶联时间缩短至7 s, 最终将单个氨基酸残基接入周期缩短至107 s. 该系统合成了包括(ALF)7 (37.5 min完成)和ACP(65-74) (17.8 min完成)等目标产物, 且粗产物纯度与传统合成法相当.
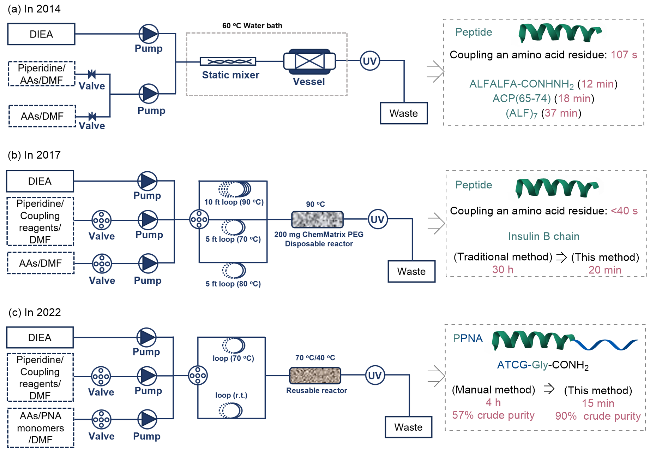
该团队[4c]在前期研究基础上进一步突破, 通过模块化设计将连续流动化学与固相合成整合, 开发出全自动化流动多肽合成(AFPS)技术, 将单个氨基酸残基的完整接入周期压缩至40 s(图16b). 该方法实现了试剂的高效混合、高温(90 ℃)活化及实时紫外监测, 不仅提高了合成速度, 还通过优化流速和温度抑制了容易外消旋化的氨基酸(如半胱氨酸和组氨酸)产生的副反应. 此外, 该技术利用紫外吸收数据动态监测Fmoc脱保护效率, 可快速识别多肽链聚集问题, 并通过调整树脂载量优化难合成序列(如JR-10肽)的产率. 与常规方法合成或商业话产品相比, AFPS在合成胰岛素B链和生长激素释放激素等长链肽时展现出相当的纯度与收率, 但耗时显著减少(如胰岛素B链合成仅需20 min, 而传统方法需30 h). 该技术通过自动化控制和连续流动反应器设计, 为精准化学合成提供了高效、可重复的平台, 适用于高通量药物候选分子库的构建和功能蛋白的快速制备.
2022年, Li等[4d]利用高效的全自动流动合成平台, 快速制备细胞穿透肽(CPP)与肽核酸(PNA)的共轭物(PPNA)(图16c). 该技术通过优化反应参数(如采用O-苯并三氮唑-四甲基脲六氟磷酸酯(HBTU)作为活化剂、70 ℃耦合温度及40 ℃去保护温度), 实现了每个酰胺键仅需10 s的高效合成, 四个肽核酸单体的PPNA粗产物纯度可达90%, 显著优于传统手动合成. 精准的温度与流速控制有效抑制了副反应(如异构体生成<1%, 碱基加合物<1%), 解决了长链PNA合成中的聚集与序列错误问题. 采用该合成技术制备的靶向IVS2-654位点的PPNA, 在剪接校正实验中活性比转染PNA提高3倍; 并且一天内可合成8条靶向SARS-CoV-2病毒5′UTR的长链PPNA. 这一技术凸显了自动化技术在复杂大分子精准合成中的有效应用.

2020年, Pentelute团队[4a]将全自动化流动多肽合成技术提升至新高度, 实现蛋白质化学合成的技术突破(图18a). 研究团队通过对反应温度、溶剂体系、活化剂类型等关键参数的精准调控, 实现了164个氨基酸长度的单域蛋白质链的连续高效合成, 其单步偶联时间仅需2.5 min, 整体合成效率较传统固相合成方法提升5倍. 尤为突出的是, 该团队通过建立多维优化体系, 可以显著抑制了天冬酰胺亚胺形成和半胱氨酸/组氨酸的外消旋化(<2%). 基于这一技术突破, 研究团队合成了9种长度在70至170个氨基酸之间的功能性蛋白链, 包括胰岛素原(86个氨基酸)和人类免疫缺陷病毒-1蛋白酶(99个氨基酸)等重要生物分子, 其合成时间均在3.5~6.5 h. 纯化后的蛋白质经折叠后表现出与重组蛋白相当的生物活性和结构稳定性, 且产物纯度更高, 所用时间更短. 另外, 该技术可灵活引入对溴苯丙氨酸等非天然氨基酸, 为功能化蛋白质设计提供了新策略. 2023年, 他 们[55]通过自动化快速流动多肽合成技术进一步实现了214个氨基酸的铜绿假单胞菌抗菌蛋白Pyocin S2 N端结构域(PyS2NTD)的“单步”精准合成, 仅耗时9.2 h, 产物纯度与生物活性均与天然蛋白相当(图18b). 近期, 该团队又完成了免疫检查点关键受体PD-1及其配体PD-L1的自动化合成[56](图18e).
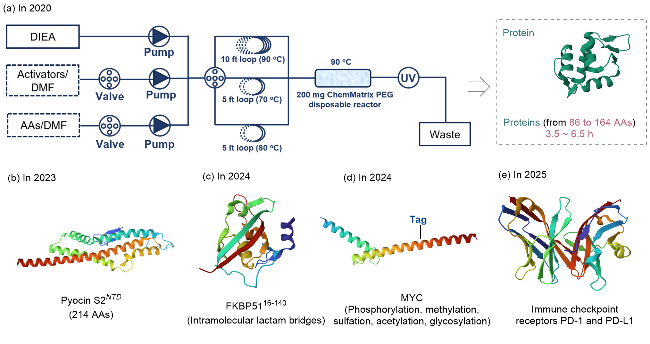
此外, Perczil和Ferentzi等[59]开发的智能多肽流动化学(SPF)系统通过精确调控流速、反应温度和压力, 实现了试剂用量(仅3 equiv.)和溶剂消耗(每循环6 mL)的降低. 针对长链多肽合成中易出现的聚集问题, 通过引入脯氨酸残基或添加溶剂(如25%二甲基亚砜/乙腈), 提高了40~80个氨基酸链段的合成纯度.
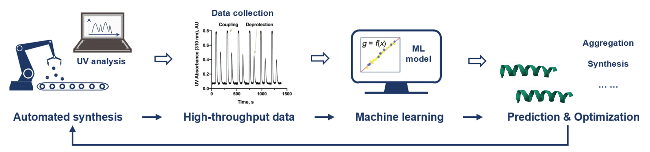
当前发展的自动化流动合成技术通过模块化设计、实时监测及高温活化等策略, 显著提高了多肽与蛋白质合成的效率、精确度及自动化程度. 这些技术已能高效制备各类复杂多肽、长链蛋白质及修饰蛋白等, 为药物研发、抗病毒治疗和功能化蛋白质设计提供了技术支撑. 然而, 面对日益复杂的蛋白质工程需求, 仍需进一步优化反应路径设计并实现合成参数的动态调控, 以解决当前生产成本较高等问题. 在这方面, 机器学习技术与自动化合成系统的融合为蛋白质精准合成领域提供了新的发展契机.
2.2 机器学习指导的自动化精准合成
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2020年, Pentelute等[61]研究了深度学习在快速流动多肽合成中的应用. 该研究通过分析35427个Fmoc脱保护反应的紫外光谱数据, 构建了一个深度学习模型, 能够以低于6%的误差预测脱保护反应的效率, 并优化多肽合成中的聚集问题. 为肽合成的实时优化和计算设计提供了新工具, 并展示了深度学习在化学合成自动化中的潜力. 随后, 该团队[62]通过自动化肽核酸合成器收集了239个Fmoc脱保护反应的实时紫外-可见数据, 并利用这些数据训练了10种不同的机器学习模型. 其中, 最佳预测准确率达到93%, 且预测的合成效率与实验测得的高效液相色谱粗产物纯度相当. 此外, 该模型成功应用于设计靶向人类抗肌萎缩蛋白基因第44号外显子、SARS-CoV-2、人类免疫缺陷以及心血管疾病、II型糖尿病和癌症相关基因的反义PNA序列, 显著提高了候选序列的筛选效率. 为高效精准的肽核酸等分子自动化合成提供了全新的智能化解决方案.
这些研究结果表明, 机器学习能够通过实时分析反应数据来优化多肽合成过程, 提升了合成效率与产物纯度. 然而, 目前数据集规模仍然有限, 需要构建更高标准的数据集, 并开发更多高效的预测模型, 同时进一步优化数据库与算法架构, 以实现更精准、更高质量的多肽/蛋白质自动化合成.
3 结论
蛋白质/多肽单一位点修饰技术与自动化合成平台的不断发展, 共同推动精准合成能力的提升, 为化学生物学、药物开发和精准医疗提供了多样化的分子工具. 本文系统总结了实现单一位点修饰的关键策略, 包括位点特异性直接修饰、配体导向修饰以及关键点定向修饰等, 并梳理了融合高温耦合、实时监测与智能控制的自动化流动合成技术的最新进展. 通过修饰靶向性与合成可控性的结合, 可实现特定单一位点修饰蛋白质/多肽的可控制备, 并大幅提升了合成的效率与精准度, 也为复杂修饰蛋白和长链蛋白质的可控合成开辟了新途径, 促进了均一化抗体-药物偶联物等新型治疗分子的开发. 然而, 现有修饰技术的普适性受到底物结构和反应条件的严格限制, 修饰位点的精准控制与反应效率仍有待提高, 一些方法在操作复杂性和成本效益等方面也有待优化. 同时, 自动化合成精度与天然蛋白质体系复杂性之间的显著差距, 以及生产成本较高等问题, 仍是制约其工业化应用的主要瓶颈. 未来, 蛋白质/多肽的精准合成仍面临若干关键挑战, 包括开发兼具高选择性高效能广适用性及操作简便性的新型修饰技术; 实现复杂修饰蛋白的高纯度规模化生产以降低成本并满足工业应用需求. 通过新型生物正交反应机制的不断突破, 以及自动化合成平台与人工智能技术的协同创新, 将会显著提升蛋白质/多肽药物的精准设计能力与临床转化潜力, 有望为重大疾病的精准干预开辟新的治疗途径.
(Lu, Y.)