


1 Introduction
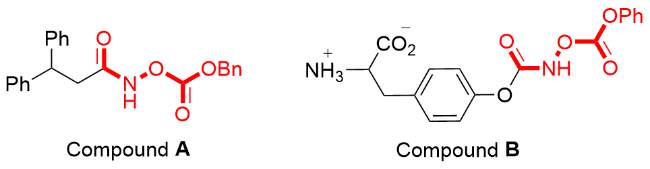
The hydroxamates are present widely in pharmaceuticals and biological activity inhibitors. Two representative examples are shown in Figure 1. Compound A was found to be the most potent β-lactamase inhibitor, with an IC50 of 0.1 and 0.23 μmol/L using meropenem and cefazolin as substrates.[1] Compound B shows significant inhibitory activity against the P99 β-lactamase (0.15 μmol/L).[2] The synthesis of hydroxamates is relatively mature due to their significant biological activity. They have been mainly prepared by reacting an acid with hydroxylamine to form an oxime under activation conditions, and then reacting with acyl chloride (Scheme 1, 1).[3] These reactions are limited by the reaction conditions and toxicity of byproducts. In addition, various types of hydroxamates are biological activity inhi- bitors. Therefore, it is desirable to develop environmentally friendly synthesis methods for new hydroxamates.
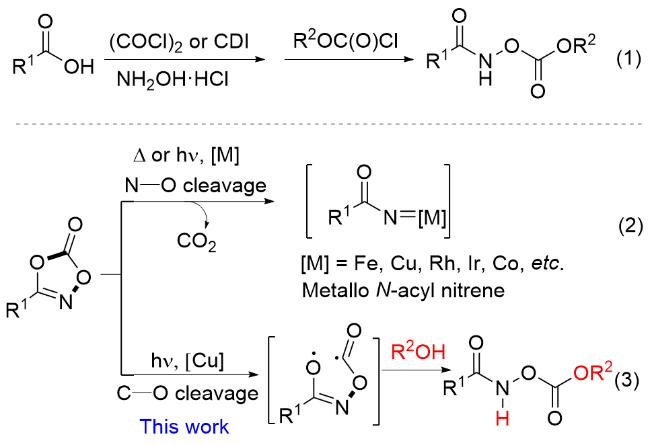
The dioxazolones are relatively stable acylation reagents, and the coupling of sp3-C—N and sp2-C—N, N—N, N—S has been reported.[4] In these works, dioxazolones underwent N—O bond cleavage under heating or light conditions, releasing CO2. The resulting intermediate then formed metal N-carbenes with transition metals. These metals included Fe, Cu, Rh, Ir, Co, etc. (Scheme 1, 2). However, the synthesis of hydroxamates with dioxazolones has no reports.
In recent years, there has been significant progress in visible light driven organic reactions. The research mainly focuses on environmental protection and green chemical synthesis. The use of inexpensive visible light sources to facilitate the synthesis of small functional organic molecules has become a powerful strategy in organic chemistry.[5] Herein, we report a new C—O bond cleavage driven by visible-light, after which reaction with alcohol generates hydroxamates based on dioxazolones (Scheme 1, 3).
2 Results and discussion
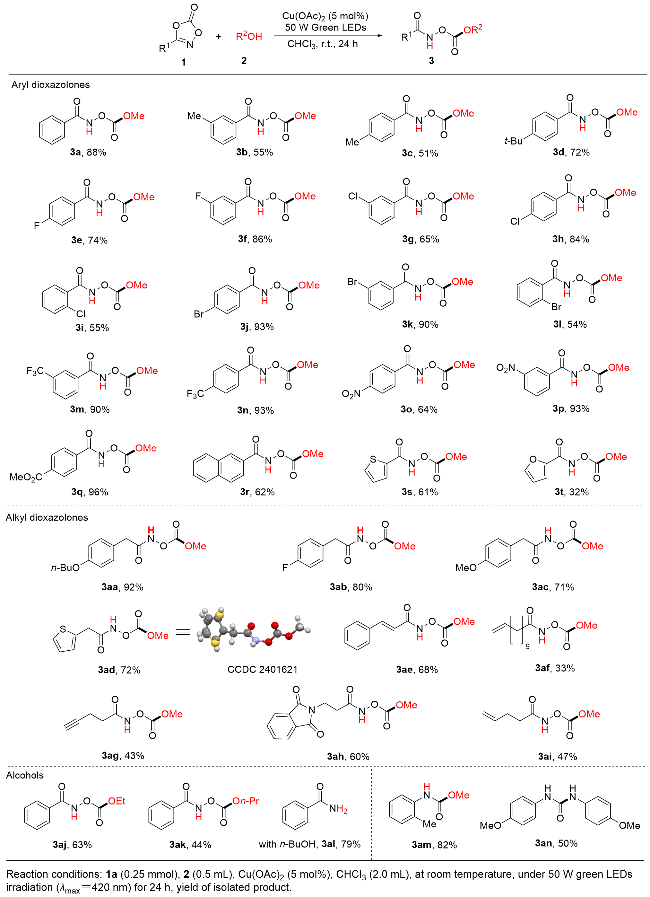
In the initial study, dioxazolone 1a and MeOH were selected as starting materials to explore the reaction parameters. The desired product 3a was obtained in 88% isolated yield in the presence of MeOH (0.5 mL), Cu(OAc)2 (5 mol%), 50 W green LEDs in CHCl3 at room temperature (Table 1, Entry 1). The product 3a was identified by mass spectrometry (MS), nuclear magnetic resonance spectroscopy (NMR) spectra. To investigate the role of light and Cu(OAc)2 in this reaction, control experiments were performed. The product 3a was not obtained without light, and was obtained in 61% isolated yield without Cu(OAc)2 (Table 1, Entries 2, 3). To investigate the impact of copper salts, CuBr•Me2S, Cu(MeCN)4PF6, CuCl were used as catalysts, providing 13%~34% isolated yields (Table 1, Entries 4~6). No desired product was observed when CuCN or CuI was used (Table 1, Entry 7). The solvent also played a crucial role. When 1,2-dichloroethane (DCE) and dichloromethane (DCM) were used as solvent, providing 37%~64% isolated yields (Table 1, Entries 8~9). To study the impact of light, the purple, blue and white light were used, respectively. We obtained the optimized reaction conditions: MeOH (0.5 mL), Cu(OAc)2 (5 mol%), 50 W green LEDs in CHCl3 at room temperature.
Table 1 Screening of reaction conditionsa |
| Entry | Light | Cu catalyst | Solvent | Yieldb/% |
|---|---|---|---|---|
| 1 | Green | Cu(OAc)2 | CHCl3 | 88 |
| 2 | Green | No | CHCl3 | 61 |
| 3 | No | Cu(OAc)2 | CHCl3 | No |
| 4 | Green | CuBr•Me2S | CHCl3 | 26 |
| 5 | Green | Cu(MeCN)4PF6 | CHCl3 | 34 |
| 6 | Green | CuCl | CHCl3 | 13 |
| 7 | Green | CuCN | CHCl3 | Trace |
| 8 | Green | CuI | CHCl3 | Trace |
| 9 | Green | Cu(OAc)2 | CH2Cl2 | 64 |
| 10 | Green | Cu(OAc)2 | ClCH2CH2Cl | 37 |
| 11 | Purple | Cu(OAc)2 | CHCl3 | 78 |
| 12 | Blue | Cu(OAc)2 | CHCl3 | 71 |
| 13 | White | Cu(OAc)2 | CHCl3 | 65 |
a Reaction conditions: 1a (0.25 mmol), MeOH (0.5 mL), Cat. (5 mol%), solvent (2.0 mL) at room temperature under irradiation with a 50 W LEDs for 24 h; b Yield of isolated product. |
With the optimized reaction conditions in hand (Table 1, Entry 1), the scope of substrates for this transformation was investigated. A series of representative dioxazolones were tested (Scheme 2). This reaction possessed good functional group compatibility and yield. We first focused on investigation of different aryl dioxazolones. The scope of substrates was then evaluated by decorating the arene rings with an array of substituents. The results demonstrated that the reaction was influenced by electron-withdrawing, electron-donating groups and steric hindrance. With electron donating, moderate yields were obtained (3b~3d, 3r), along with the formation of 50% urea 3an, while with electron-withdrawing, better yields were obtained (3f~3h, 3j~3k, 3m~3q)). In addition, this reaction was significantly affected by steric hindrance, and gave a moderate yield (3i~3l). When substrate of 2-Me was used, only the rearrangement product was obtained (3am, 82%). This reaction could also tolerate heterocyclic ring, and had a moderate yield (3s, 3t, 3ad). Secondly, a range of the alkyl dioxazolones was investigated. To our delight, the reaction could provide good yields (3aa~3ai) for active functional groups, such as double bonds, triple bonds, and amides (3ae~3ai), which is beneficial for subsequent functionalization research. Lastly, the applicability of alcohols was studied. Ethanol and propanol could provide desired products. However, when n-BuOH was used, the reduction product was observed (3al). Surprisingly, the raw material was obtained when i-PrOH, t-BuOH and BnOH were used.
To understand the reaction mechanism, a series of control experiments were carried out (Scheme 3). To determine the involvement of any free radical intermediate in the reaction, various known radical quenchers [1,1,5,5-tetramethylpen- tamethylene nitroxide (TEMPO), 3,5-di-tert-4-butylhydr-oxytoluene (BHT), 1,1-diphenylethylene] were added, respectively (Scheme 3, i).[6] The results showed that TEMPO had a significant impact on the isolated yield. Compound 3ak was obtained in 65% isolated yield, and 3ao was detected by high-resolution magnetic resonance spectroscopy (HMRS). When BHT was added, the product was not observed. However, 1,1-diphenylethylene had no significant effect on the isolated yield. From the results, the reaction might be free radical reaction process. To determine whether there was an extrication and absorption process of CO2 in the reaction, PPh3 was used to capture the N-carbene product (Scheme 3, ii).[7] As a result, 3an could not be observed under standard conditions. 3an was obtained under conditions: PPh3 (3.0 equiv.), Cu(OAc)2 (20 mol%), 80 ℃, DCE (Scheme 3, iii). The deuterium substitution experiments were employed to observe the source of amide hydrogen. The results showed that amide hydrogen might come from MeOH via 1H NMR analysis of the product (Scheme 3, iv~v). Next, the deuterium kinetic isotope effect (KIE) of the dioxazolone was measured in parallel of MeOH and MeOD. The resultant KIE was kH/kD=1.2∶1 on basis of the isolated yields (Scheme 3, vi). The data showed that the cleavage of O—H bond in MeOH was not likely involved in the rate-limiting step. When irradiated, the solution containing dioxazolone 1a, MeOH and Cu- (OAc)2, a distinct electron paramagnetic resonance (EPR) signal from CuII was detected (Figure 2a). Free radical production was detected in the dark and in the light for 5 min (Figure 2b). The light on-off irradiation experiments revealed that the reaction was facilitated by light, no conversion was observed when the reaction mixture was stirred in the dark, indicating the necessity for continuous irradiation (Figure 2c). Next, ultraviolet-visible light experiments were performed with dioxazolone 1a, MeOH, Cu(OAc)2, and CHCl3. While no obvious changes occurred in the absence of either compound 1a or MeOH in CHCl3, a clear absorption was observed when dioxazolone 1a, MeOH and Cu(OAc)2 were mixed in CHCl3. The results indicate that Cu(OAc)2 plays a facilitating role in this reaction (Figure 2d).
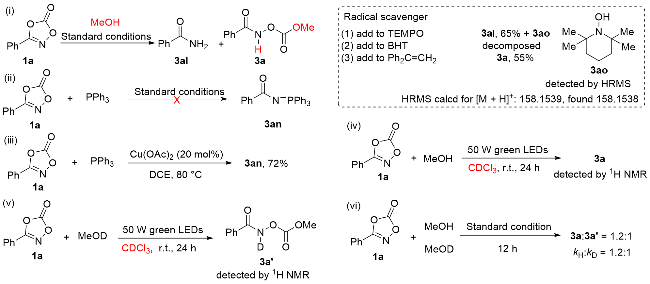
Scheme 3 Mechanistic studies(i) Experiment of radical capture; (ii) and (iii) experiment of nitrogen capture; (iv) and (v) deuterium exchange experiment; (vi) KIE experiment |
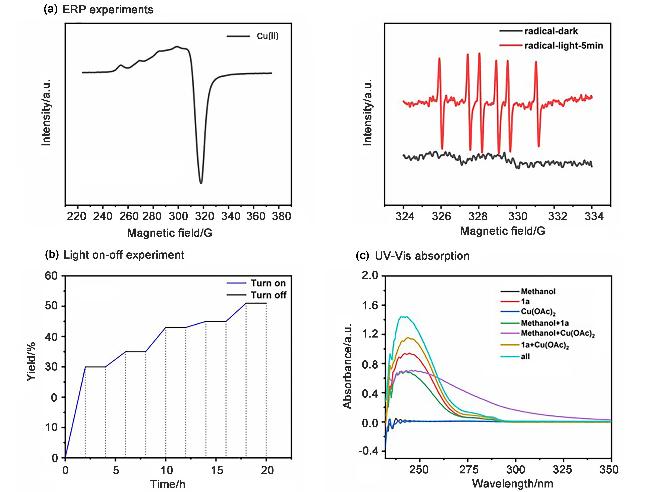
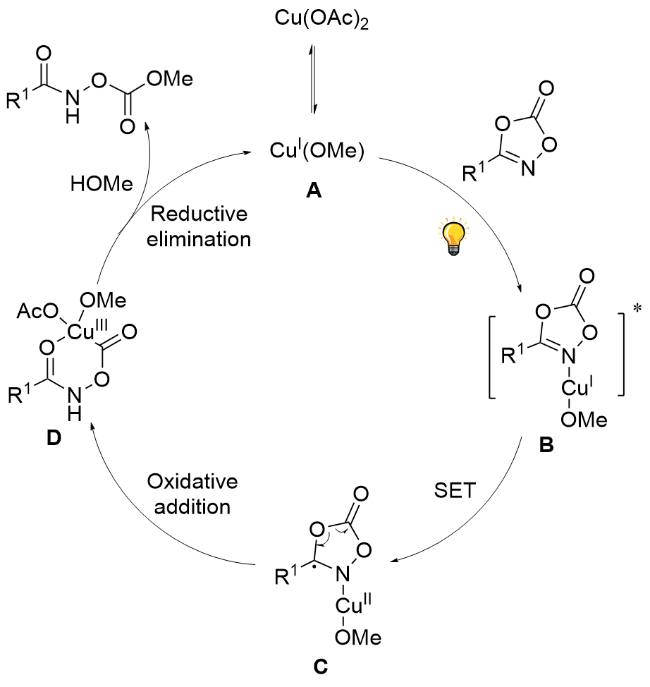
A probable mechanism was proposed (Scheme 4) based on the preliminary experiments and the literature reports.[8,7a,9] The reaction may proceed through CuII salt which coordinates with MeOH to form complex A and then coordinates with dioxazolone 1 to generate B under in visible-light-driven. Subsequently, the cleavage of C—O leads to the formation of intermediate C via binding with hydrogen free radicals. Consequently, the complex C generates D via an oxidation addition process. Ultimately, inter- mediate D generates 3 and complex A via a reductive elimination with MeOH.
To inspect the applicability of the reaction, we attempted to broaden scale synthesis of 3a (Scheme 5). Dioxazolone 1a was transformed into the expected product in the broadening scale in 85% isolated yield.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 Conclusions
We have developed an efficient copper-catalyzed synthesis of hydroxamates with dioxazolones under mild reaction conditions, which was driven by visible light. The method tolerated a variety of functional groups with good to excellent yields. According to experimental observation, the reaction might be free radical reaction process and the H free radical might be generated.
4 Experimental section
4.1 General information
All commercially reactants don’t need further purification. The ultra dry solvents were used as commercially supplied, such as tetrahydrofuran (THF), CH2Cl2 and MeOH etc. Flash chromatography was performed on silica gel (200~300 mesh). 1H NMR spectra were recorded at 400 MHz on a JNM-ECZ400S (JEOL, Japan) spectrometer. Chemical shifts (δ) are referenced to the appropriate residual solvent pea. 13C NMR spectra were recorded at 100 MHz on a JNM-ECZ400S (JEOL, Japan) spectrometer. Chemical shifts (δ C) are referenced to the appropriate residual solvent peak. 19F NMR spectra were recorded at 376 or 471 MHz on a JNM-ECZ400S (JEOL, Japan) spectrometer and quoted to 1 decimal place and with coupling constants (J) to the nearest 0.1 Hz. Low resolution and high resolution mass spectra were recorded on a DIONEX Ulti- Mate 300 & Bruker ESI-TOF Compact spectrometer.
4.2 General procedure for the reaction of dioxazolones with alcochols
Compounds 1a~1s, 1aa~1ah (0.25 mmol) and Cu(OAc)2 (2.3 mg, 0.013 mmol, 0.05 equiv.) were dissolved in CHCl3 (2 mL), and 2 (0.5 mL) was added. The mixture was stirred at room temperature under irradiation with a 50 W green LEDs for 24 h. The solution was concentrated and purified by column chromatography.
N-((Methoxycarbonyl)oxy)benzamide (3a): Yellow solid, yield 88% (42.1 mg), Rf=0.34 (CH2Cl2), m.p. 36~37 ℃; 1H NMR (400 MHz, CDCl3) δ: 10.77 (s, 1H), 7.79~7.75 (m, 2H), 7.47 (t, J=7.3 Hz, 1H), 7.34 (t, J=7.7 Hz, 2H), 3.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.9, 155.2, 132.8, 130.3, 128.8, 127.7, 56.5. HRMS (ESI) calcd for C9H9NO4 [M+H]+ 195.0609, found 195.0610.
N-((Methoxycarbonyl)oxy)-3-methylbenzamide (3b): Yellow oil, yield 55% (25.9 mg), Rf=0.36 (CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 9.69 (s, 1H), 7.61 (s, 1H), 7.58 (d, J=7.5 Hz, 1H), 7.34 (d, J=7.7 Hz, 1H), 7.29 (t, J=7.5 Hz, 1H), 3.91 (s, 3H), 2.35 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 167.2, 155.5, 138.9, 133.7, 130.3, 128.8, 128.3, 124.6, 56.6, 21.4. HRMS (ESI) calcd for C10H11NO4 [M+H]+ 210.0766, found 210.0767.
N-((Methoxycarbonyl)oxy)-4-methylbenzamide (3c): Yellow oil, yield 51% (24.1 mg), Rf=0.35 (CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 9.64 (s, 1H), 7.69 (d, J=8.2 Hz, 2H), 7.21 (d, J=7.9 Hz, 2H), 3.91 (s, 3H), 2.38 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 167.1, 155.6, 143.7, 129.5, 127.6, 127.5, 56.6, 21.7. HRMS (ESI) calcd for C10H11NO4 [M+H]+ 210.0768, found 210.0769.
4-(tert-Butyl)-N-((methoxycarbonyl)oxy)benzamide (3d): Yellow solid, yield 72% (33.0 mg), Rf=0.41 (CH2Cl2), m.p. 91~92 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.48 (s, 1H), 7.75 (d, J=8.5 Hz, 2H), 7.45 (d, J=8.7 Hz, 2H), 3.93 (s, 3H), 1.32 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 167.1, 156.7, 155.6, 127.5, 125.9, 56.6, 35.2, 31.2. HRMS (ESI) calcd for C13H17NO4 [M+H]+ 252.1227, found 252.1252.
4-Fluoro-N-((methoxycarbonyl)oxy)benzamide (3e): Ye- llow solid, yield 74% (34.8 mg), Rf=0.32 (CH2Cl2), m.p. 45~46 ℃; 1H NMR (400 MHz, CDCl3) δ: 10.02 (s, 1H), 7.81 (dd, J=8.8, 5.3 Hz, 2H), 7.08 (t, J=8.7 Hz, 2H), 3.91 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.0, 165.6 (d, J=235 Hz), 155.4, 130.2 (d, J=10 Hz), 126.5 (d, J=3 Hz), 116.1 (d, J=22 Hz), 56.7; 19F NMR (376 MHz, CDCl3) δ: -115.2 (s, 1F). HRMS (ESI) calcd for C9H8FNO4 [M+ H]+ 214.0515, found 214.0519.
3-Fluoro-N-((methoxycarbonyl)oxy)benzamide (3f): Ye- llow oil, yield 86% (40.1 mg), Rf=0.34 (CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 10.06 (s, 1H), 7.57 (d, J=7.6 Hz, 1H), 7.50 (d, J=9.3 Hz, 1H), 7.39 (dd, J=13.5, 7.9 Hz, 1H), 7.23 (t, J=8.3 Hz, 1H), 3.91 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 165.6, 162.7 (d, J=247 Hz), 155.2, 132.3, 130.7 (d, J=8 Hz), 123.2 (d, J=4 Hz), 120.0 (d, J=21 Hz), 115 (d, J=23 Hz), 56.7; 19F NMR (376 MHz, CDCl3) δ: -111.0 (s, 1F). HRMS (ESI) calcd for C9H8FNO4 [M+H]+ 214.0512, found 214.0516.
3-Chloro-N-((methoxycarbonyl)oxy)benzamide (3g): Yellow oil, yield 55% (25.6 mg), Rf=0.35 (CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 9.89 (s, 1H), 7.78 (s, 1H), 7.66 (d, J=7.8 Hz, 1H), 7.51 (d, J=8.0 Hz, 1H), 7.36 (t, J=7.9 Hz, 1H), 3.93 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 165.6, 155.3, 135.1, 133.1, 132.0, 130.2, 127.9, 125.7, 56.7. HRMS (ESI) calcd for C9H8ClNO4 [M+H]+ 230.0220, found 230.0221.
4-Chloro-N-((methoxycarbonyl)oxy)benzamide (3h): Yellow solid, yield 84% (39.1 mg), Rf=0.34 (CH2Cl2), m.p. 97~98 ℃; 1H NMR (400 MHz, CDCl3) δ: 10.36 (s, 1H), 7.70 (d, J=8.6 Hz, 2H), 7.35 (d, J=8.5 Hz, 2H), 3.88 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.0, 155.3, 139.3, 129.1, 129.1, 128.6, 56.7. HRMS (ESI) calcd for C9H8Cl- NO4 [M+H]+ 230.0221, found 230.0222.
2-Chloro-N-((methoxycarbonyl)oxy)benzamide (3i): Brown oil, yield 55% (25.6 mg), Rf=0.35 (CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 9.54 (s, 1H), 7.70 (d, J=7.8 Hz, 1H), 7.44 (d, J=1.3 Hz, 1H), 7.42 (d, J=1.1 Hz, 1H), 7.35 (dd, J=7.7, 5.0 Hz, 1H), 3.95 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 164.7, 155.0, 132.7, 131.5, 130.9, 130.8, 130.6, 127.3, 56.7. HRMS (ESI) calcd for C9H8ClNO4 [M+H]+ 230.0221, found 230.0233.
4-Bromo-N-((methoxycarbonyl)oxy)benzamide (3j): Ye- llow solid, yield 93% (42.1 mg), Rf=0.40 (CH2Cl2), m.p. 90~91 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.75 (s, 1H), 7.65 (d, J=8.6 Hz, 2H), 7.57 (d, J=8.6 Hz, 2H), 3.93 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.1, 155.4, 132.2, 129.2, 129.1, 128.0, 56.7. HRMS (ESI) calcd for C9H8Br- NO4 [M+H]+ 273.9710, found 273.9714.
3-Bromo-N-((methoxycarbonyl)oxy)benzamide (3k): Yellow solid, yield 90% (40.8 mg), Rf=0.42 (CH2Cl2), m.p. 55~56 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.91 (s, 1H), 7.70 (d, J=7.8 Hz, 1H), 7.63 (d, J=8.1 Hz, 1H), 7.26 (t, J=8.0 Hz, 1H), 3.89 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 165.2, 155.1, 135.8, 132.1, 130.8, 130.4, 126.2, 122.9, 56.7. HRMS (ESI) calcd for C9H8BrNO4 [M+H]+ 273.9711, found 273.9715.
2-Bromo-N-((methoxycarbonyl)oxy)benzamide (3l): Ye- llow oil, yield 60% (27.2 mg), Rf=0.41 (CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 9.33 (s, 1H), 7.63 (d, J=1.2 Hz, 1H), 7.61 (d, J=1.5 Hz, 1H), 7.41~7.32 (m, 2H), 3.95 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 165.5, 155.5, 133.8, 133.3, 132.7, 130.5, 127.7, 120.1, 56.7. HRMS (ESI) calcd for C9H8BrNO4 [M+H]+ 273.9714, found 273.9717.
N-((Methoxycarbonyl)oxy)-3-(trifluoromethyl)benzami-de (3m): Yellow solid, yield 90% (38.3 mg), Rf=0.30 (CH2Cl2), m.p. 58~59 ℃; 1H NMR (400 MHz, CDCl3) δ: 10.16 (s, 1H), 8.06 (s, 1H), 7.98 (d, J=7.8 Hz, 1H), 7.78 (d, J=7.8 Hz, 1H), 7.56 (t, J=7.8 Hz, 1H), 3.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 164.3, 155.2, 131.3 (t, J=33 Hz), 130.9, 129.6, 129.5 (d, J=3 Hz), 124.9, 124.7, 122.2 (d, J=3 Hz), 56.9; 19F NMR (376 MHz, CDCl3) δ: -62.9 (s, 3F). HRMS (ESI) calcd for C10H8F3NO4 [M+H]+ 264.0481, found 264.0483.
N-((Methoxycarbonyl)oxy)-4-(trifluoromethyl)benzami-de (3n): White solid, yield 93% (39.6 mg), Rf=0.35 (CH2Cl2), m.p. 110~111 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.01 (d, J=8.2 Hz, 2H), 7.92 (d, J=8.2 Hz, 2H), 3.89 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 164.0, 155.0, 134.9, 132.6 (d, J=32 Hz), 128.8, 126.3 (d, J=4 Hz), 124.2 (d, J=271 Hz), 56.9; 19F NMR (376 MHz, CDCl3) δ: -63.1 (s, 3F). HRMS (ESI) calcd for C10H8F3- NO4 [M+H]+ 264.0483, found 264.0486.
N-((Methoxycarbonyl)oxy)-4-nitrobenzamide (3o): Ye- llow solid, yield 64% (29.5 mg), Rf=0.29 (CH2Cl2), m.p. 89~90 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.38 (d, J=9.1 Hz, 2H), 8.05 (d, J=8.9 Hz, 2H), 3.90 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 163.6, 155.0, 150.3, 136.7, 129.5, 124.5, 57.0. HRMS (ESI) calcd for C9H8N2O6 [M+H]+ 241.0461, found 241.0467.
N-((Methoxycarbonyl)oxy)-3-nitrobenzamide (3p): Whi- te solid, yield 93% (42.9 mg), Rf=0.35 (CH2Cl2), m.p. 142~143 ℃; 1H NMR (400 MHz, Acetone-d6) δ: 11.99 (s, 1H), 8.69 (s, 1H), 8.50 (d, J=8.2 Hz, 1H), 8.32 (d, J=7.8 Hz, 1H), 7.88 (t, J=8.0 Hz, 1H), 3.94 (s, 3H); 13C NMR (100 MHz, Acetone-d6) δ: 163.3, 154.9, 148.5, 133.6, 132.7, 130.6, 127.0, 122.2, 56.0. HRMS (ESI) calcd for C9H8N2O6 [M+H]+ 241.0464, found 241.0468.
Methyl 4-((methoxycarbonyl)oxy)carbamoyl)benzoate (3q): Yellow solid, yield 96% (43.9 mg), Rf=0.30 (CH2Cl2), m.p. 118~119 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.89 (s, 1H), 8.06 (d, J=8.8 Hz, 2H), 7.85 (d, J=8.3 Hz, 2H), 3.93 (s, 6H); 13C NMR (100 MHz, CDCl3) δ: 166.3, 166.0, 155.3, 134.3, 133.9, 130.1, 127.8, 56.8, 52.7. HRMS (ESI) calcd for C11H11NO6 [M+H]+ 254.0665, found 254.0666.
N-((Methoxycarbonyl)oxy)-2-naphthamide (3r): Yellow solid, yield 62% (28.5 mg), Rf=0.35 (CH2Cl2), m.p. 84~85 ℃; 1H NMR (400 MHz, CDCl3) δ: 10.04 (s, 1H), 8.31 (s, 1H), 7.82 (d, J=6.9 Hz, 2H), 7.79 (s, 2H), 7.52 (dd, J=16.9, 8.2 Hz, 2H), 3.91 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 167.2, 155.6, 135.4, 132.4, 129.2, 128.8, 128.8, 128.4, 127.9, 127.5, 127.0, 123.5, 56.6. HRMS (ESI) calcd for C13H11NO4 [M+H]+ 246.0728, found 246.0734.
N-((Methoxycarbonyl)oxy)thiophene-2-carboxamide(3s): Brown oil, yield 61% (29.0 mg), Rf=0.55 (CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 9.95 (s, 1H), 7.69 (d, J=2.4 Hz, 1H), 7.56 (d, J=3.7 Hz, 1H), 7.09~7.05 (m, 1H), 3.90 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 162.0, 155.5, 133.4, 132.2, 130.7, 128.1, 56.7. HRMS (ESI) calcd for C7H7NO4S [M+H]+ 201.0128, found 201.0122.
N-((Methoxycarbonyl)oxy)furan-2-carboxamide (3t): Brown oil, yield 32% (15.5 mg), Rf=0.45 (CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 9.61 (s, 1H), 7.49 (dd, J=1.7, 0.8 Hz, 1H), 7.25 (dd, J=3.6, 0.8 Hz, 1H), 6.53 (dd, J=3.6, 1.7 Hz, 1H), 3.93 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 157.2, 155.3, 145.4, 144.7, 117.3, 112.4, 56.7. HRMS (ESI) calcd for C7H7NO5 [M+H]+ 186.0402, found 186.0403.
2-(4-Butoxyphenyl)-N-((methoxycarbonyl)oxyacetami-de (3aa): Yellow solid, yield 92% (41.8 mg), Rf=0.55 (CH2Cl2), m.p. 86~87 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.52 (s, 1H), 7.20 (d, J=8.6 Hz, 2H), 6.88 (d, J=8.7 Hz, 2H), 3.94 (t, J=6.6 Hz, 2H), 3.89 (s, 3H), 3.60 (s, 2H), 1.79~1.72 (m, 2H), 1.48 (dd, J=15.1, 7.4 Hz, 2H), 0.97 (t, J=7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 158.9, 155.1, 130.6, 124.3, 115.2, 67.8, 56.6, 40.0, 31.4, 19.3, 13.9. HRMS (ESI) calcd for C14H19NO5 [M+H]+ 287.1754, found 287.1750.
2-(4-Fluorophenyl)-N-((methoxycarbonyl)oxy)acetami-de (3ab): White solid, yield 80% (37.3 mg), Rf=0.35 (CH2Cl2), m.p. 42~43 ℃; 1H NMR (400 MHz, Acetone-d6) δ: 11.10 (s, 1H), 7.36 (dd, J=8.6, 5.5 Hz, 2H), 7.08 (t, J=8.9 Hz, 2H), 3.84 (s, 3H), 3.57 (s, 2H); 13C NMR (100MHz, Acetone-d6) δ: 168.4, 162.0 (d, J=242 Hz), 155.1, 131.2 (d, J=8 Hz), 115.1 (d, J=21 Hz), 55.8, 38.1. 19F NMR (376 MHz, CDCl3) δ: -117.5 (s, 3F). HRMS (ESI) calcd for C14H19NO5 [M+H]+ 227.1904, found 227.1902.
N-((Methoxycarbonyl)oxy)-2-(4-methoxyphenyl)aceta-mide (3ac): Brown solid, yield 72% (33.8 mg), Rf=0.43 (CH2Cl2), m.p. 95~96 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.72 (s, 1H), 7.22 (d, J=5.2 Hz, 1H), 6.97 (m, 1H), 6.95 (d, J=3.3 Hz, 1H), 3.86 (s, 3H), 3.80 (s, 2H); 13C NMR (100 MHz, CDCl3) δ: 168.2, 154.9, 134.0, 127.7, 127.4, 125.8, 56.6, 34.2. HRMS (ESI) calcd for C8H9NO4S [M+H]+ 216.0311, found 216.0305.
N-((Methoxycarbonyl)oxy)-2-(thiophen-2-yl)acetamide(3ad): White solid, yield 71% (41.6 mg), Rf=0.50 (CH2Cl2/ EtOAc, V∶V=10∶1), m.p. 59~60 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.19 (s, 1H), 7.20 (d, J=8.7 Hz, 2H), 6.86 (d, J=8.7 Hz, 2H), 3.86 (s, 3H), 3.78 (s, 3H), 3.54 (s, 2H); 13C NMR (100 MHz, CDCl3) δ: 169.8, 159.1, 155.1, 130.6, 124.9, 114.5, 56.5, 55.4, 39.5. HRMS (ESI) calcd for C11H13NO5 [M+H]+ 239.2311, found 239.2310.
N-((Methoxycarbonyl)oxy)cinnamamide (3ae): Yellow solid, yield 68% (31.8 mg), Rf=0.53 (CH2Cl2), m.p. 120~121 ℃; 1H NMR (400 MHz, CDCl3) δ: 10.23 (s, 1H), 7.75 (d, J=15.6 Hz, 1H), 7.49~7.46 (m, 2H), 7.32 (d, J=6.7 Hz, 3H), 6.56 (d, J=15.5 Hz, 1H), 3.87 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 165.7, 155.3, 144.2, 134.3, 130.5, 129.0, 128.3, 115.3, 56.6. HRMS (ESI) calcd for C11H11N- O4 [M+H]+ 222.0766, found 222.0769.
N-((Methoxycarbonyl)oxy)dodec-11-enamide (3af): Ye- llow oil, yield 33% (15.1 mg), Rf=0.52 (CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 9.08 (s, 1H), 5.85~5.74 (m, 1H), 5.00~4.90 (m, 2H), 3.90 (s, 3H), 2.23 (t, J=7.4 Hz, 2H), 2.05~1.99 (m, 2H), 1.70~1.62 (m, 2H), 1.37~1.24 (m, 10H); 13C NMR (100 MHz, CDCl3) δ: 172.0, 155.3, 139.3, 114.3, 56.5, 33.9, 32.8, 29.3, 29.3, 29.1, 29.1, 29.0, 25.1. HRMS (ESI) calcd for C13H23NO4 [M+H]+ 258.1741, found 258.1738.
N-((Methoxycarbonyl)oxy)pent-4-ynamide (3ag): Yellow solid, yield 21% (10.3 mg), Rf=0.53 (CH2Cl2), m.p. 83~84 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.38 (s, 1H), 3.92 (s, 3H), 2.56 (t, J=3.2 Hz, 2H), 2.50 (t, J=6.8 Hz, 2H), 2.04 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 169.8, 155.1, 82.2, 70.0, 56.6, 32.0, 14.4. HRMS (ESI) calcd for C7H9NO4 [M+H]+ 172.0698, found 172.0601.
3-(1,3-Dioxoisoindolin-2-yl)-N-((methoxycarbonyl)-oxy)propanamide (3ah): Yellow solid, yield 60% (27.0 mg), Rf=0.50 (EtOAc/CH2Cl2, V∶V=1∶4), m.p. 130~131 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.63 (s, 1H), 7.82 (dd, J=5.7, 3.1 Hz, 2H), 7.70 (dd, J=5.4, 3.0 Hz, 2H), 4.04 (t, J=7.1 Hz, 2H), 3.86 (s, 3H), 2.74 (t, J=7.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 168.3, 155.0, 134.3, 132.0, 123.5, 56.6, 33.7, 317, 29.8. HRMS (ESI) calcd for C13H12- N2O6 [M+H]+ 293.0771, found 293.0774.
N-((Methoxycarbonyl)oxy)pent-4-enamide (3ai): Yellow oil, yield 42% (20.6 mg), Rf=0.42 (CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 9.44 (s, 1H), 5.87~5.77 (m, 1H), 5.04 (t, J=9.0 Hz, 2H), 3.90 (s, 3H), 2.44~2.39 (m, 2H), 2.34 (t, J=7.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 171.1, 155.2, 136.3, 116.2, 56.5, 32.1, 28.9. HRMS (ESI) calcd for C7H11NO4 [M+H]+ 173.0702, found 173.0703.
N-((Ethoxycarbonyl)oxy)benzamide (3aj): Brown oil, yield 63% (32.3 mg), Rf=0.50 (CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 9.50 (s, 1H), 7.81~7.79 (m, 2H), 7.54 (t, J=7.3 Hz, 1H), 7.45~7.41 (m, 2H), 4.33 (q, J=7.2 Hz, 2H), 1.36 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.9, 154.8, 132.9, 130.4, 128.9, 127.6, 66.5, 14.2. HRMS (ESI) calcd for C10H11NO4 [M+H]+ 210.0795, found 210.0701.
N-((Propoxycarbonyl)oxy)benzamide (3ak): Yellow oil, yield 44% (24.1 mg), Rf=0.35 (CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 9.49 (s, 1H), 7.81~7.78 (m, 2H), 7.55 (t, J=7.5 Hz, 1H), 7.43 (t, J=7.9 Hz, 2H), 4.24 (t, J=6.7 Hz, 2H), 1.80~1.71 (m, 2H), 0.98 (t, J=7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 167.0, 155.0, 133.0, 130.5, 128.9, 127.6, 72.0, 22.0, 10.1. HRMS (ESI) calcd for C11H13NO4 [M+H]+ 224.0923, found 224.0924.
Benzamide (3al): White solid, yield 79% (23.6 mg), Rf=0.50 (EtOAc/CH2Cl2, V∶V=1∶1), m.p. 129~130 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.84~7.80 (m, 2H), 7.52 (t, J=7.4 Hz, 1H), 7.44 (t, J=7.4 Hz, 2H), 6.32 (s, 2H); 13C NMR (100 MHz, CDCl3) δ: 169.9, 133.5, 132.1, 128.7, 127.5. HRMS (ESI) calcd for C7H7NO [M+H]+ 121.0598, found 121.1200.
Methyl o-tolylcarbamate (3am): Yellow solid, yield 82% (42.3 mg), Rf=0.35 (CH2Cl2), m.p. 45~46 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.76 (s, 1H), 7.23~7.16 (m, 2H), 7.04 (t, J=6.3 Hz, 1H), 6.49 (s, 1H), 3.78 (s, 3H), 2.25 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 154.6, 135.9, 130.5, 127.0, 124.4, 52.5, 17.7. HRMS (ESI) calcd for C10H11NO4 [M+H]+ 210.0767, found 210.0768.
1,3-Bis(4-methoxyphenyl)urea (3an): Yellow solid, yield 50% (24.1 mg), Rf=0.35 (CH2Cl2), m.p. 64~65 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.38 (s, 2H), 7.34 (d, J=9.2 Hz, 4H), 6.86 (d, J=9.2 Hz, 4H), 3.71 (s, 6H); 13C NMR (100 MHz, DMSO-d6) δ: 154.8, 153.5, 133.5, 120.4, 114.5, 55.7. HRMS (ESI) calcd for C15H16N2O3 [M+H]+ 272.1298, found 272.3001.
Supporting Information Crystallographic data, 1H NMR and 13C NMR spectra of compounds 3a~3t and 3aa~3an. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
(Lu, Y.)