

1 Introduction
Cycloheximide, a well-known glutarimide-containing polyketide natural product, is widely utilized as a protein translation inhibitor in biotechnology research.[1] It serves as a valuable tool for studying diverse biological processes, including cell apoptosis mechanisms,[2] protein expression regulation,[3] cell cycle dynamics, pathological model design, animal learning and memory functions.[4] Actiphenol, which shares the core carbon skeleton of cycloheximide but replaces the cyclohexanone moiety with a phenol group, exhibits promising bioactivities such as antioxidant effects and inhibition of cell migration.[5-6] Research indicates that both cycloheximide and actiphenol are biosynthesized through a shared pathway.[7] Elucidating the structural features and biological properties of new glutarimide derivatives could expand our understanding of this pharmacologically important compound class and its biosynthetic versatility.
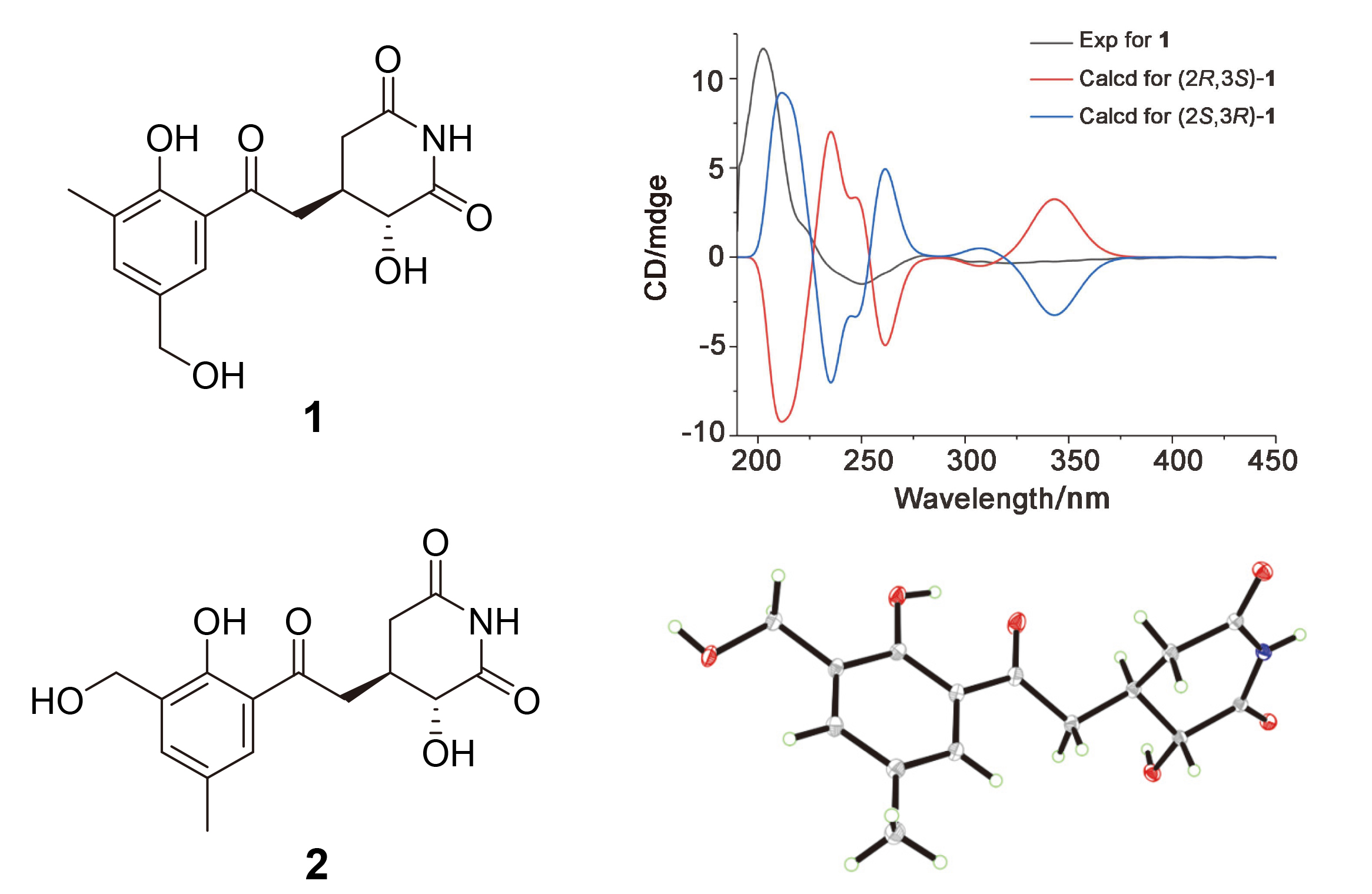
In the continuous pursuit of novel bioactive microbial metabolites, we have developed an integrated discovery platform combining genome mining, antibiotic resistance profiling, and antimicrobial activity screening to identify promising endophytic actinomycetes from medicinal plants,[8-9] which led to the identification of Streptomyces sp. YINM00048 as a particularly productive strain. This endophytic actinomycete was originally isolated from Agrimonia pilosa Ledeb. collected in the biodiversity-rich Gaoligong Mountains region of Yunnan Province, China. Through systematic isolation of the ethyl acetate extract from large-scale fermentation, 10 purified compounds (Figure 1) were obtained, including two new actiphenol analogues, 2-hydroxy-3-[2-(2-hydroxy-3-methylphenyl-5-hydroxymethyl)-2-oxoethyl] glutarimide (1) and 2-hydro-xy-3-[2-(2-hydroxy-3-hydroxymethyl-5-methylphenyl)-2-oxoethyl] glutarimide (2), together with eight known glutarimide derivatives (3~10). Herein, the isolation, structure elucidation, absolute configuration assignment and bioactivity of these metabolites were described in detail.
2 Results and discussion
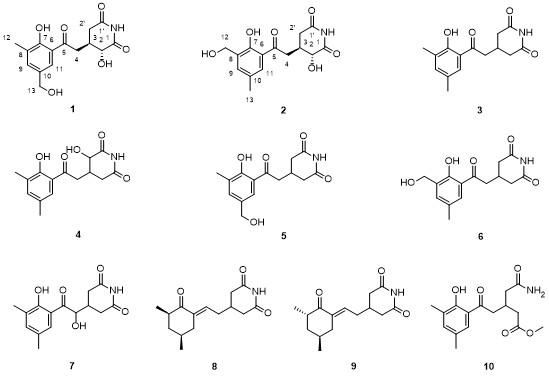
Compound 1 has an outward appearance of pale yellow solid. HR-ESI-MS analysis (m/z 306.0990 [M-H]-, calcd for C15H16NO6 306.0983) of 1 revealed its molecular formula as C15H17NO6, containing eight degrees of unsaturation. Analysis of the 1H NMR spectrum (Table 1) revealed the presence of one methyl, three methylenes, along with four methine protons. The 13C NMR and distortionless enhancement by polarization transfer (DEPT) indicated the presence of fifteen carbon resonances, which contained seven non-protonated carbons (δC 206.5, 176.1, 173.8, 161.2, 132.8, 128.4, and 119.6), four methines (δC 137.6, 127.5, 72.1, and 35.4), three methylenes (δC 64.6, 41.0, and 37.4), and a methyl (δC 15.5). Structural characterization of compound 1 was achieved through detailed NMR analysis and comparison with the known analog 3-[2-[2-hydroxy-3-methylphenyl-5-(hydroxymethyl)]-2-oxoethyl] glutarimide (5).[5] Through systematic NMR analysis (Figure 2A), it is first established that compound 1 shares the same carbon skeleton with 5 but features significant structural modification. Definitive heteronuclear multiple bond correlation spectroscopy (HMBC) correlations (H-9/C-7, C-11, C-12, C-13; H-11/C-5, C-7, C-9, C-13; H-12/C-7, C-8, C-9; H- 13/C-9, C-10, C-11) unambiguously confirmed the presence of a 1,2,3,5-tetrasubstituted benzene ring, while the continuous 1H-1H correlation spectroscopy (COSY) coupling network [H-2 (H-2')/H-3/H-4] along with characteristic HMBC correlations (H-2/C-1, C-3, C-4; H2'/C-1', C-2, C-3, C-4; H-3/C-4, C-5; H-4/C-2, C-3, C-5; H-11/ C-5] collectively verified the glutarimide ring system fused with the benzene moiety. Particularly noteworthy was the significant downfield shift of C-2 (δC 72.3, Δ+35.3) in compound 1 compared to 5. This observation, combined with the molecular formula analysis, conclusively demonstrated the hydroxyl substitution at C-2. Based on these multidimensional evidences, compound 1 was unequivocally identified as 2-hydroxy-3-[2-(2-hydroxy-3-methyl- phenyl-5-hydroxymethyl)-2-oxoethyl] glutarimide, representing a novel C-2 hydroxylated derivative of compound 5.
Table 1 1H NMR (400 MHz) and 13C NMR (101 MHz) data of compound 1 in CD3OD |
| Position | δC, type | δH, mult (J in Hz) |
|---|---|---|
| 1 | 176.1, C | |
| 1′ | 173.8, C | |
| 2 | 72.1, CH | 4.18, d (11.5) |
| 2′ | 37.4, CH2 | 2.56, dd (17.2, 12.3) |
| 2.87, dd (17.2, 4.2) | ||
| 3 | 35.4, CH | 2.72, m |
| 4 | 41.0, CH2 | 3.09, dd (17.3, 8.8) |
| 3.66, dd (17.4, 3.5) | ||
| 5 | 206.5, C | |
| 6 | 119.6, C | |
| 7 | 161.2,.C | |
| 8 | 128.4, C | |
| 9 | 137.6, CH | 7.40, s |
| 10 | 132.8, C | |
| 11 | 127.5, CH | 7.74, s |
| 12 | 15.5, CH3 | 2.23, s |
| 13 | 64.6, CH2 | 4.54, s |
The relative configuration of 1 was elucidated through comprehensive analysis of ROESY correlation. Critical through-space interaction between H-2 and H-4 consistently supported a 2,3-trans relative configuration. To determine the absolute stereochemistry, the time-dependent density functional theory (TDDFT) calculations of electronic circular dichroism (ECD) spectra were performed. The superior agreement between experimental and calculated ECD curves (Figure 2B) unambiguously assigned the absolute configuration as 2S,3R.
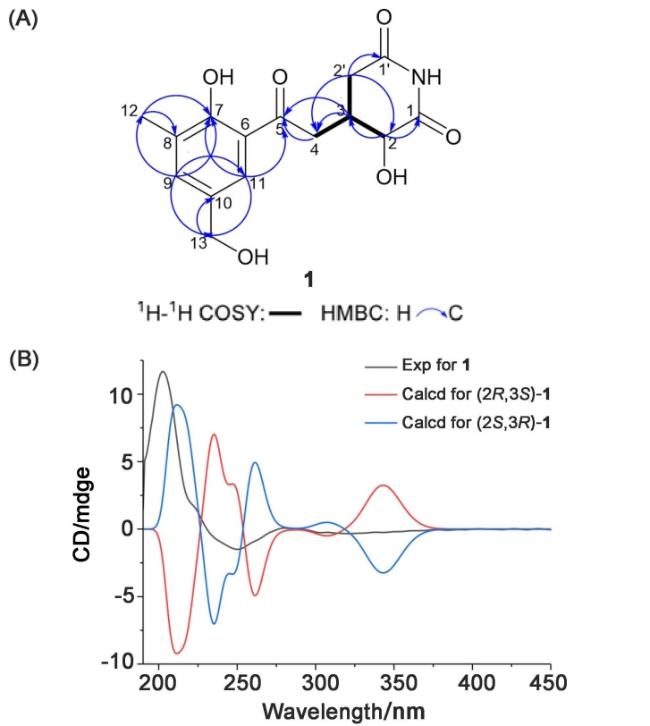
Compound 2 was isolated as pale yellow crystals. HR- ESI-MS analysis displayed a protonated molecular ion peak at m/z 308.1128 [M+H]+ (calcd for C15H18NO6 308.1129), establishing the molecular formula as C15H17NO6 with eight degrees of unsaturation. The 1H NMR spectroscopic data for 2 (Table 2) presented a singlet methyl, four methine protons. Comprehensive analysis of 13C NMR and DEPT spectra identified fifteen carbon resonances categorized as seven non-protonated carbons (δC 205.8, 174.5, 171.9, 156.4, 130.7, 127.3, 118.4), four methines (δC 134.9, 128.4, 70.4, 33.7), three methylenes (δC 57.3, 40.2, 36.1), and one methyl (δC 20.3). Comparative NMR analysis revealed that compound 2 shares the identical carbon framework with the known compound 6,[12] but also exhibits a significant downfield shift of C-2 (δC 70.8, Δ+33.8), strongly suggesting hydroxylation at this position. This modification was further confirmed by key HMBC correlations between H-2/C-3 and H-4/C-2, C-3 (Figure 3A). Meanwhile, detailed comparison of the NMR spectra with compound 1 demonstrated that these two analogs differ exclusively in their benzene ring substitution patterns, as evidenced by distinct chemical shifts at C-12 and C-13 (Tables 1 and 2). These collective spectroscopic data unambiguously established the planar structure of 2 as depicted in Figure 3A.
Table 2 1H NMR (400 MHz) and 13C NMR (101 MHz) data of compound 2 in DMSO-d6 |
| Position | δC, type | δH, mult (J in Hz) |
|---|---|---|
| 1 | 174.5, C | |
| 1′ | 171.9, C | |
| 2 | 70.4, CH | 4.05, d (9.6) |
| 2′ | 36.1, CH2 | 2.59, m |
| 3 | 33.7, CH | 2.54, m |
| 4 | 40.2, CH2 | 3.04, dd (17.1, 7.5) |
| 3.47, m | ||
| 5 | 205.8, C | |
| 6 | 118.4, C | |
| 7 | 156.4, C | |
| 8 | 130.7, C | |
| 9 | 134.9, CH | 7.48, s |
| 10 | 127.3, C | |
| 11 | 128.4, CH | 7.63, s |
| 12 | 57.3, CH2 | 4.51, s |
| 13 | 20.3, CH3 | 2.29, s |
| OH-7 | 12.23, s | |
| NH | 10.83, s |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The stereochemical configuration of 2 was comprehensively characterized through multiple analytical techniques. Initial ROESY analysis revealed key correlation between H-2 and H-4, indicating spatial proximity and supporting a trans relative configuration. This assignment was also confirmed by single-crystal X-ray diffraction analysis (Figure 3C, CCDC 2301876), which not only verified the planar structure but also determined the absolute configuration to be 2S,3R. Furthermore, the absolute configuration of 2 was verified through TDDFT calculations of ECD spectra. The close correspondence between experimental and calculated ECD spectra (Figure 3B) provided independent confirmation of the 2S,3R configuration.
Structural elucidation of compounds 3~10 was achie- ved through comprehensive spectroscopic comparison with literature data. The known compounds were unambiguously identified as actiphenol (3),[10] 4-hydroxy-3-[5-dimethylphenyl)-2-oxoethyl] glutarimide (4),[11] 3-[2-(2-hydroxy-3-methylphenyl-5-hydroxymeth- yl)-2-oxoethyl] glutarimide (5),[5] 3-[2-(2-hydroxy-3-hy- droxymethyl-5-methylphenyl)-2-oxoethyl] glutarimide (6),[12] nongkang 101 (7),[13] 4-[2′-(3″(R),5″(S)-3″,5″-dime- thyl-2″-oxylcyclohexylidene (8),[14] 4-[2′-(3″(R),5″(S)-3″, 5″-dimethyl-2″-oxylcyclohexylidene (9),[14] methyl 5- amino-3-(2-(2-hydroxy-3,5-dimethylphenyl)-2-oxoethyl)- 5-oxopentanoate (10).[15]
To evaluate the antitumor potential of the new compounds 1 and 2, their cytotoxic activities were systematically assessed against five human cancer cell lines representing diverse tumor types: HL-60 (leukemia), A549 (non-small cell lung cancer), SMMC-7721 (hepatocellular carcinoma), MCF-7 (breast adenocarcinoma), and SW480 (colorectal adenocarcinoma). Initial screening at 40 μmol/ L demonstrated distinct biological profiles among the tested compounds. While compound 1 showed negligible growth inhibition across all cell lines (<30% inhibition at 40 μmol/L), compound 2 displayed selective cytotoxicity, particularly against A549 and SMMC-7721 cells at this concentration. Dose-response analysis revealed that 2 exhibited weak but consistent cytotoxic activity against A549 and SMMC-7721 cells with IC50 values of (27.51±1.13) and (31.12±1.93) μmol/L (Table 3), respectively.
Table 3 Anti-tumor activity of compound 2 |
| Compd. | IC50±SD/(μmol•L-1) | |
|---|---|---|
| A549 | SMMC-7721 | |
| 2 | 27.51±1.13 | 31.12±1.93 |
| DDP | 13.78±1.07 | 8.668±0.153 |
| Taxol | <0.008 | 0.063±0.010 |
3 Conclusions
Two glutarimide derivatives were discovered and characterized from an endophytic Streptomyces sp. YINM00048 associated with Agrimonia pilosa Ledeb. Structural characterization demonstrated that compounds 1 and 2 are polyhydroxy-substituted actiphenol derivatives containing modified benzene and glutarimide moieties. Biological evaluation revealed that compound 2 exhibited weak cytotoxic activity against human lung carcinoma (A549) and hepatoma (SMMC-7721) cell lines, with IC50 values of 62.69 and 55.07 μmol/L, respectively. These discoveries not only substantially enrich the structural diversity of glutarimide-containing natural products, particularly within the cycloheximide-actiphenol family, but also underscore the remarkable biosynthetic potential of plant- associated endophytic actinomycetes as valuable sources of novel bioactive metabolites.
4 Experimental section
4.1 General experimental procedures
X-ray data were collected on a Bruker Apex DUO diffractometer (Bruker Corporation, Germany). Optical rotations were recorded in MeOH using an Autopol IV polarimeter (Rudolph, America). Circular dichroism (CD) spectra were recorded on a Chirascan V100 spectropolarimeter (Applied Photophysics Ltd., Surrey, UK). UV spectra were acquired in MeOH with a Shimadzu UV-2550 PC spectrometer (Shimadzu Co., Ltd, Tokyo, Japan). Infrared (IR) spectra were obtained with a Nicolet NEXUS 470 spectrophotometer in KBr discs (Thermo Fisher Scientific, USA). NMR spectra were obtained on a Bruker Avance-400 MHz instrument (Bruker, Karlsruhe, Germany) using TMS as an internal standard. HR-ESI-MS data were obtained using an Agilent 1100 Q-TOF mass instrument (Agilent, Santa Clara, CA, United States). Column chromatography (CC) was performed using silica gel (200~300 mesh, Qingdao Marine Chemical Group Co., Qingdao, China), Sephadex LH-20 (GE Healthcare Bio-Science AB, Uppsala, Sweden) and Lichroprep RP-18 gel (40~63 μm, Merck, Darmstadt, Germany). Thinlayer chromatography (TLC) was performed using silica gel GF254 plates (Qingdao Haiyang Chemical Co., Ltd, Qingdao, China) with various solvent systems. Semipreparative HPLC was conducted on an Agilent 1260 series equipped with a DAD detector and a Zorbax SB-C18 (250 mm×4.6 mm, 5 μm) column.
4.2 Strain material
The strain was isolated from the Agrimonia pilosa Ledeb. collected at Gaoligong Mountains region of Yunnan Province, China, in October 2021. It was identified as a Streptomyces griseus subsp. griseus strain by morphological examination and gene sequence analysis. The 16S rRNA gene sequence of Streptomyces sp. YINM00048 was deposited in GenBank under accession number PV759728, and the voucher specimen was deposited at School of Medicine, Yunnan University.
4.3 Fermentation, extraction and isolation
The strain Streptomyces sp. YINM00048 was grown on medium ISP2 agar plates (yeast extract 4 g, malt extract 10 g, glucose 4 g, agar 18 g in 1 L water, pH 7.0) for 3~5 days at 28 ℃. Then, proper amounts of spores of this strain were inoculated into 250 mL baffled Erlenmeyer flasks containing 100 mL of the sterile seed medium (medium ISP2: yeast extract 4 g/L, malt extract 10 g/L, glucose 4 g/L, pH 7.0) and cultivated for 48 h at 28 ℃ on a rotary shaker (200 rpm). After that, aliquots (5 mL) of the seed culture were transferred into 500 mL baffled Erlenmeyer flasks filled with 100 mL of sterile production medium (medium 20#: glycerol 50 g/L, corn meal 25 g/L, nutritonal yeast seasoning 5g/L, pH 7.0), and cultured on a rotary shaker (200 r/min) at 28 ℃ for 15 days. The fermentation broth (27.5 L) was extracted three times with equal volume of ethyl acetate. The ethyl acetate extract was subsequently evaporated to afford 21.89 g creamy crude extract.
The crude extract (21.89 g) was subjected to silica gel column chromatography (CC) eluted stepwise with dichloromethane-methanol (V∶V=1∶0, 50∶1, 30∶1, 10∶1) to afford four fractions (Fr.A~Fr.D) on TLC analysis. Fr.A was chromatographed on a Sephadex LH-20 column and eluted with dichloromethane-methanol (V∶ V=1∶1) to give two subfractions (Fr.A-1 and Fr.A-2). Fr.A-1 was then purified by semi-preparative HPLC (ZORBAX SB-C18, 250 mm×9.4 mm, 5 μm; solvent A, water; solvent B, methanol; 70%~80% B, 0~25 min; flow rate at 3.0 mL/min) to obtain compound 3 (6.5 mg, tR=21.25 min). Fr.A-2 was eluted stepwise with MeOH-H2O (V∶ V=1∶10~0∶1) by LiChroprep RP-18 column and further purified by semi-preparative HPLC (ZORBAX SB- C18, 250 mm×9.4 mm, 5 μm; solvent A, water; solvent B, methanol; 70%~85% B, 0~20 min; flow rate at 3.0 mL/ min) to obtain compound 10 (3.2 mg, tR=12.64 min). Fr.B was eluted with dichloromethane-methanol (V∶V=1∶1) on a Sephadex LH-20 column to give two subfractions (Fr.B-1 and Fr.B-2). Fr.B-1 was then separated by semi- preparative HPLC (ZORBAX SB-C18, 250 mm×9.4 mm, 5 μm; solvent A, water; solvent B, methanol; 70%~80% B, 0~15 min; flow rate at 3.0 mL/min) to obtain compound 4 (5.8 mg, tR=9.58 min). Fr.B-2 was isolated by semi-pre- parative HPLC (ZORBAX SB-C18, 250 mm×9.4 mm, 5 μm; solvent A, water; solvent B, methanol; 50%~75% B, 0~30 min; flow rate at 3.0 mL/min) to afford compounds 6 (3.9 mg, tR=10.74 min), 7 (3.5 mg, tR=16.63 min), 8 (2.5 mg, tR=17.41 min), 9 (2.6 mg, tR=16.18 min). Finally, Fr.C was chromatographed on a Sephadex LH-20 column and eluted with methanol to give two subfractions (Fr.C-1 and Fr.C-2). Fr.C-1 was further separated by semi-prepara- tive HPLC (ZORBAX SB-C18, 250 mm×9.4 mm, 5 μm; solvent A, water; solvent B, methanol; 45%~70% B, 0~20 min; flow rate at 3.0 mL/min) to obtain compound 2 (3.8 mg, tR=8.92 min). Fr.C-2 was eluted stepwise with MeOH- H2O (V∶V=0∶1~7∶20) on a LiChroprep RP-18 column, and further isolated by semipreparative HPLC (ZORBAX SB-C18, 250 mm×9.4 mm, 5 μm; solvent A, water; solvent B, methanol; 40%~65% B, 0~30 min; flow rate at 3.0 mL/min) to afford compounds 1 (3.6 mg, tR=15.25 min), and 5 (3.8 mg, tR=24.04 min).
2-Hydroxy-3-[2-(2-hydroxy-3-methylphenyl-5-hydro-xymethyl)-2-oxoethyl] glutarimide (1): Pale yellow solid; $[\alpha]_{\mathrm{D}}^{25}$+16.37 (c 0.10, MeOH); UV (MeOH) λmax [log ε/(L•mol-1•cm-1)]: 219 (2.78), 260 (2.45), 342 (1.77) nm; IR (KBr) νmax: 3482, 1723, 1690 cm-1. HR-ESI-MS calcd for C15H16NO6 [M-H]- 306.0983, found 306.0990. 1H NMR and 13C NMR data see Table 1.
2-Hydroxy-3-[2-(2-hydroxy-3-hydroxymethyl-5-meth-ylphenyl)-2-oxoethyl] glutarimide (2): Pale yellow crystalline solid; $[\alpha]_{\mathrm{D}}^{25}$+19.60 (c 0.10, MeOH); UV (MeOH) λmax [log ε/(L•mol-1•cm-1)]: 259 (2.62), 345 (1.53) nm; IR (KBr) νmax: 3426, 1738, 1685 cm-1. HR-ESI-MS calcd for C15H18NO6 308.1129 [M+H]+, found 308.1128. 1H NMR and 13C NMR data see Table 2.
4.4 Crystallographic data of 2
Suitable crystals of 2 were obtained from a mixed solvent of MeOH-H2O (V∶V=30∶1). Compound 2: C15H17NO6, M=307.29, a=0.57296(2) nm, b=1.65653(5) nm, c=2.87404(9) nm, α=90°, β=90°, γ=90°, V=2.72782(15) nm3, T=100.00 K, space group P212121, Z=8, μ(Cu Kα)=0.984 mm-1, 54692 reflections measured, 5815 independent reflections (Rint=0.0362). The final R1 values were 0.0269 (I>2σ(I)) and 0.0275 (all data), while the final wR(F2) values were 0.0708 (I>2σ(I)) and 0.0714 (all data). The goodness of fit on F2 was 1.027, and the flack parameter was -0.02(4) (CCDC No. 2301876).
4.5 TDDFT-ECD calculations
Starting from the conformation deduced from the ROESY spectrum and Chem3D modeling, conformational searches were initially performed using CONFLEX version 7.0 with the Merck molecular force field (MMFF) 94 s force-field 5 kcal/mol above the ground state.[16] Density functional theory (DFT) calculations were used to generate and optimize the conformers. For each conformer, ECD spectra were computed using TDDFT at the B3LYP/6- 31+G(d,p)//B3LYP/6-31G(d) theoretical level in Gaussian 09,[17-18] and the calculated ECD spectra of the different conformers were simulated with a half bandwidth of ca. 0.3 eV. Solvent effects were considered using the polarizable continuum model (PCM). The calculated ECD spectrum was generated by Boltzmann-weighted summation of individual conformer spectra, processed using SpecDis 1.64 software. The calculated ECD curves were corrected by UV and compared to the experimental data.[19]
4.6 Cytotoxicity assay
The in vitro cytotoxic activities of the compounds were evaluated against five human cancer cell lines (HL-60, SMMC-7721, A-549, MCF-7, and SW480) using the MTS assay.[20] Following ATCC protocols, cells were plated in 96-well microplates (3×10² cells/well) with 100 μL Roswell Park Memorial Institute (RPMI) medium and cultured at 37 ℃ under 5% CO2 for 24 h. After incubation, methyl trichlorosilane (MTS) reagent (Promega, Madison, WI, USA) was introduced and the absorbance at 490 nm recorded using a microplate reader. IC50 values were determined through Reed and Muench's method, with cisplatin serving as positive control.[21] Results represent mean± SD values from triplicate determinations.
Supporting Information NMR and HR-ESI-MS spectra for new compounds 1 and 2. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
(Lu, Y.)