

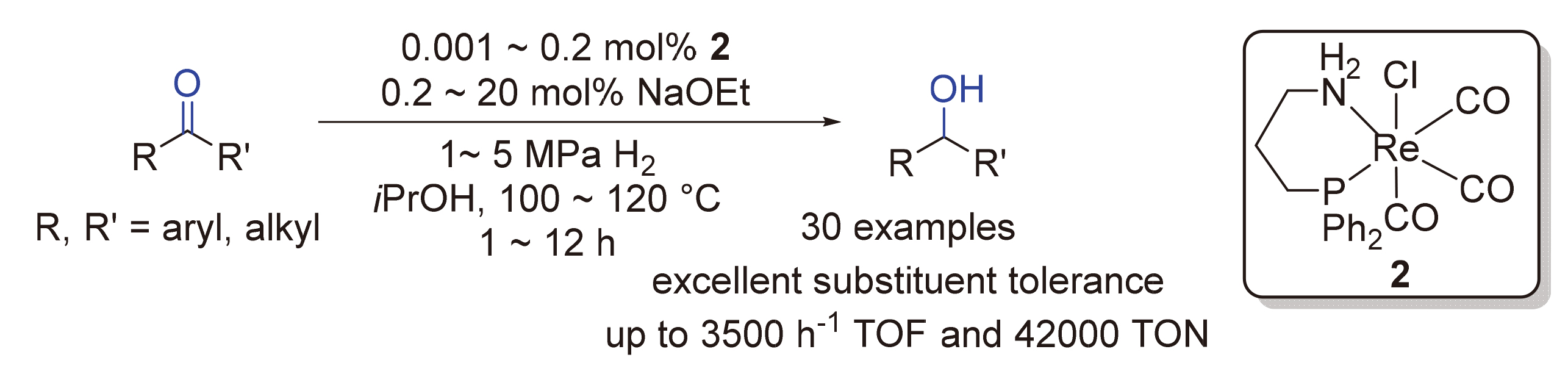
自1995年Noyori等[1]报道金属-NH配体协同催化以来, 基于金属氢(M-H)与配体胺氢(N-H)协同作用配合物已发展成为均相加氢领域最具代表性催化剂体系之一[2]. 截至目前, 围绕贵金属钌[3]、锇[4]、铱[5]以及廉价金属铁[6]、钴[7]和锰[8]等, 研究人员成功研制出一系列具有不同结构特征的金属-NH配合物, 取得了显著的研究进展. 第VII族过渡金属铼, 作为当前多相催化加氢领域的“明星”金属之一, 受到广泛关注[9]. 然而, 在均相加氢研究中, 围绕铼构建配合物催化剂的研究尚处于发展初期[10]. 如图1所示, 目前文献中仅有几例基于NH配体协同作用铼配合物应用于催化加氢的报道, 且活性停留在较低的水平(转化数<1000)[11]. 此外, 结构较为单一, 均是由三齿螯合型PNP配体构成. 众所周知, 配体的结构对其所构成配合物催化性能有着决定性的影响. 通过采用不同类型膦胺配体, 制备结构多样的Re(I)-NH配合物并考察其催化加氢性能, 提升铼在均相加氢领域的应用水平, 是值得开展的研究课题.
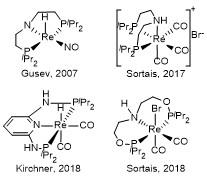
近来, 我们课题组构筑出一类配位有四齿PNNP配体的铼配合物, 并成功应用于醛、酮等羰基衍生物分子加氢反应中[12]. 活性测试结果显示, PNNP-Re(I)催化剂活性相比文献报道有显著提升, 催化苯乙酮加氢转化数(TON)最高达到61008. 这一结果充分展现出Re(I)-NH配合物应用于催化加氢的潜力. 相比螯合型配体, 双齿PN配体结构较为简单, 且其所构成的钌[3a,13]、锰[8b]等配合物已成功应用于酮、酯等羰基衍生物分子加氢反应研究中. 2021年, Wass等[14]报道合成PN-Re(I)配合物(Ph2PCH2CH2NH2)Re(CO)3Br、[(Ph2PCH2CH2NH2)2Re-(CO)2]Br和[(Ph2PCH2CH2CH2NH2)2Re(CO)2]Br, 并将其成功应用于催化甲醇和乙醇转化生成异丁醇的研究. 基于我们前期在PN-Ru(II)催化剂研究领域所取得的进展[15], 本工作系统考察了PN-Re(I)配合物催化加氢反应性能. 围绕配体Ph2P(CH2)2NH2、Ph2P(CH2)3NH2和o-Ph2PC6H4NH2, 成功制备出金属中心分别配位有一个或两个PN配体的铼配合物1~6. 在对配合物结构进行系统表征基础上考察了催化苯乙酮加氢制1-苯乙醇反应活性, 并围绕2探究了催化剂的底物适用性.
1 结果与讨论
1.1 铼配合物合成及结构表征
铼配合物1~6结构如图2所示. 与Wass等[14]采用Re(CO)5Br为金属前驱盐制备PN-Re(I)配合物相似, Re(CO)5Cl与1 equiv. Ph2P(CH2)2NH2(或Ph2P(CH2)3-NH2、o-Ph2PC6H4NH2)配体的甲苯溶液在100 ℃条件下反应24 h, PN配体取代两个CO生成配合物(Ph2PCH2-CH2NH2)Re(CO)3Cl (1)、(Ph2PCH2CH2CH2NH2)Re-(CO)3Cl (2)和(o-Ph2PC6H4NH2)Re(CO)3Cl (3). 有趣的是, 以1,4-二氧六环为溶剂, Re(CO)5Cl与2 equiv. PN配体在110 ℃件下反应2 d, 生成的是金属中心分别配位有两个PN配体和两个CO的配合物[(Ph2PCH2CH2-NH2)2Re(CO)2]Cl (4)、[(Ph2PCH2CH2CH2NH2)2Re(CO)2]- Cl (5)和[(o-Ph2PC6H4NH2)2Re(CO)2]Cl (6). 与1(或2、3)结构中含有Re—Cl共价键不同, 配合物4(或5、6)金属中心配位饱和, 以离子化合物形式存在.
分别对配合物1~6进行了核磁共振、红外光谱和高分辨质谱等表征, 确定了分子组成和结构. 以CDCl3或DMSO-d6为氘代试剂, 测得1~3的31P NMR谱图中化学位移分别在δ 30.57、-4.13以及28.23处显示出一个单峰, 表明1~3在溶液中均以单一构型存在. 与1相似, 4~6的31P NMR谱化学位移也分别仅在δ 40.20、-3.25、40.65处显示出一个共振信号, 说明4~6结构中两个PN配体化学环境相同, 在溶液中以高度对称的分子构型存在. 相比配合物1(和3、4、6), 推测是由于胺基对二苯基膦基所造成的影响不同, 配合物2和5的31P NMR谱信号均向高场偏移[16]. 配合物1~3以及4~6的13C NMR谱图中分别显示出3个(1: δ 190.42、191.77、195.44; 2: δ 190.88、191.41、194.79; 3: δ 189.92、193.09、196.39)和1个(4: δ 199.93; 5: 200.31; 6: 200.40)羰基碳共振吸收峰, 以及红外光谱图中分别显示出3个(1: 2020、1920、1880 cm-1; 2: 2010、1920、1860 cm-1; 3: 2018、1910、1889 cm-1)和2个(4: 1912、1815 cm-1; 5: 1910、1810 cm-1; 6: 1930、1840 cm-1) C—O伸缩振动吸收峰, 进一步验证了上述分子构型.
1.2 铼配合物活性评价
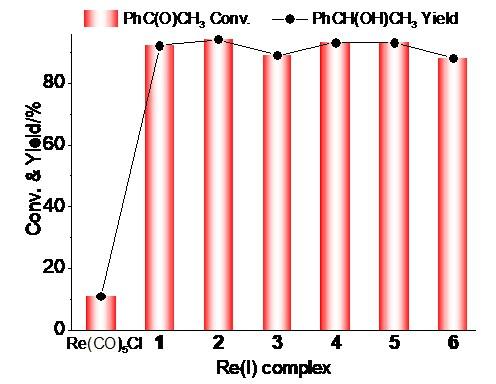
在100 ℃、初始H2压力2 MPa、4 h、铼配合物以及助剂NaOEt用量分别为0.1和10 mol%等条件下, 以催化苯乙酮加氢制1-苯乙醇为模型反应, 考察了1~6的催化加氢性能. 如图4所示, 相比刚性o-Ph2P-C6H4NH2配体所构成铼配合物3和6, 分别取得89%和88%收率, 围绕柔性PN配体构建的1、2、4和5均取得>90%的收率, 其中以2最优(94%). 配合物3和6活性相对较低的原因推测是, 由于o-Ph2PC6H4NH2配体的刚性结构阻碍了底物分子与活性中心的有效接触[15b]. 值得注意的是, 在相同反应条件下, Re(CO)5Cl仅取得11%的收率, 凸显出PN配体的不可或缺性. 与配体结构对催化剂性能存在一定影响不同, 金属中心配位的PN配体数量对PN-Re(I)配合物加氢活性基本没有影响. 这一结果与PN-Mn(I)催化剂体系中, 第二PN配体引入对加氢性能造成不利影响的实验现象不同[8b], 推测是得益于Re(I)较Mn(I)离子半径大、位阻效应影响较小所致.
在分别优化得出i-PrOH和NaOEt为最佳溶剂和助剂基础上, 围绕配合物2进一步考察了温度、H2压力和助剂用量等反应条件对催化体系加氢性能的影响. 如表1所示, 在图4的反应条件下反应1 h, 1-苯乙醇的收率达80% (Entry 1); 将反应温度升高至120 ℃, 反应1 h 后产物即可获得近乎定量的收率(Entry 3). 将H2压力降至1 MPa对催化剂2的催化性能基本没有影响, 在相同反应时间内, 1-苯乙醇收率仅从94%降低至90% (Entries 2, 4). 相比H2压力, 反应温度对催化剂2的催化加氢活性的影响更为显著. 如Entries 5, 6所示, 在80 ℃以及2 MPa (或1 MPa) H2条件下反应8 h, 1-苯乙醇收率仅分别达到64%和62%.
表1 铼(I)配合物2催化苯乙酮加氢制备1-苯乙醇aTable 1 Catalytic hydrogenation of acetophenone to 1-phenyl- ethanol by 2 |

| Entry | 2/mol% | Temp./ ℃ | p(H2)/ MPa | Time/h | Yieldb/ % | TOF/ h-1 | TON |
|---|---|---|---|---|---|---|---|
| 1 | 0.1 | 100 | 2 | 1 | 80 | 803 | 803 |
| 2 | 0.1 | 100 | 2 | 4 | 94 | 236 | 942 |
| 3 | 0.1 | 120 | 2 | 1 | >99 | 999 | 999 |
| 4 | 0.1 | 100 | 1 | 4 | 90 | 226 | 904 |
| 5 | 0.1 | 80 | 2 | 8 | 64 | 80 | 636 |
| 6 | 0.1 | 80 | 1 | 8 | 62 | 78 | 624 |
| 7c | 0.1 | 100 | 2 | 4 | 10 | 26 | 104 |
| 8d | 0.1 | 100 | 2 | 4 | 52 | 130 | 518 |
| 9e | 0.1 | 100 | 2 | 4 | 81 | 203 | 812 |
| 10f | 0.01 | 100 | 5 | 12 | >99 | 833 | 9990 |
| 11g | 0.002 | 120 | 5 | 12 | 50 | 2079 | 24950 |
| 12h | 0.001 | 120 | 5 | 12 | 42 | 3500 | 42000 |
a Reaction conditions: 2.0 mmol of acetophenone (0.5 mmol/mL i-PrOH solution), 10 mol% NaOEt. b Determined by GC. c 1 mol% NaOEt. d 2 mol%. e 5 mol%. f 5.0 mmol of acetophenone, 1 mol% NaOEt. g 12.0 mmol of acetophenone, 0.4 mol% NaOEt. h 24.0 mmol of acetophenone, 0.2 mol% NaOEt. |
在Ru(II)、Fe(II)和Mn(I)等金属-NH配体协同作用催化剂体系中, 助剂“碱”在提升催化剂加氢活性方面发挥着至关重要的作用, 且在一定范围内随着碱用量的增加性能逐步提升[6d,8b,18]. 针对这一现象, 研究人员开展了广泛而深入的研究工作. 目前, 普遍接受的观点是, 反应过程中碱金属阳离子(M′)取代N-H, 生成N-M′结构基元增强M-H亲核性, 进而实现催化加氢活性的提升[19]. 有趣的是, 在当前反应体系中也存在类似的实验现象. 当NaOEt的用量为1 mol% 时, 该反应体系中1-苯乙醇的收率仅为10% (Entry 7); 进一步增加NaOEt用量至2 mol%或5 mol%, 1-苯乙醇的收率提升至52%和81% (Entries 8, 9). 综合上述结果可以推测, 当前催化剂体系也是基于金属氢和配体胺氢协同作用的催化加氢反应机制.
值得注意的是, 在较低催化剂的用量条件下2也表现出优异的加氢活性. 如Entry 10所示, 在2的用量为0.01 mol%以及5 MPa H2等条件下反应12 h, 1-苯乙醇的收率可达>99%. 在120 ℃等条件下进一步降低2的用量至0.002 mol%或0.001 mol% (S/C=50000和100000), 反应12 h后, 目标产物的收率分别为50%和42% (Entries 11, 12), TOF值和TON值最高分别达到3500 h-1和42000. 这一结果虽与我们近期报道的PNNP-Re(I)催化剂体系[12]最优水平(TOF=4041 h-1、TON=61008)还存在一定差距, 但相比前期文献中最优结果(TOF=46.5 h-1, TON=930; 0.1 mol%铼配合物[HN(CH2CH2OPi- Pr2)2]Re(CO)2Br、5 MPa H2、120 ℃)仍有70余倍和40余倍提升, 进一步展现出Re(I)-NH催化剂体系用于催化加氢的良好前景. PNNP-Re(I)催化剂体系表现出更优异催化性能的原因, 在于四齿PNNP配体更强的螯合能力既提升了催化剂的稳定性, 又能更高效地调控其电子性质[12,17,20].
1.3 底物范围的拓展
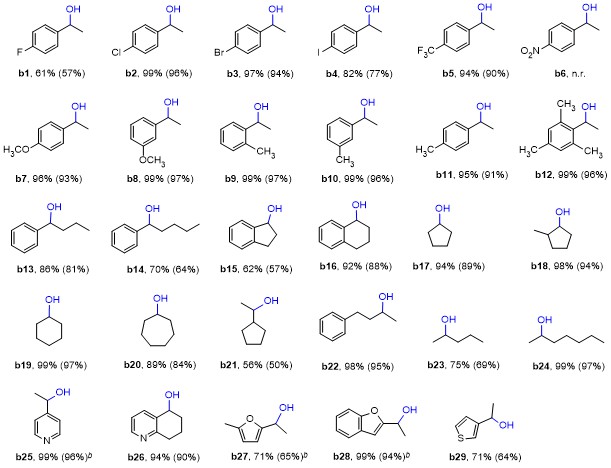
在催化剂用量0.1 mol%、反应温度100 ℃、氢气压力2 MPa、反应时间6 h的优化条件下, 考察了催化剂2对不同结构酮类分子的加氢反应性能(表2). 除对硝基苯乙酮(a6)外,催化剂2对对位含卤素(a1~a4)、吸电子基三氟甲基(a5)及给电子基甲氧基(a7)、甲基(a11)等官能团取代的苯乙酮进行加氢反应时, 均表现出一定的加氢活性, 对应醇类产物的收率为57%~96%. 与催化剂PNNP-Re(I)难以有效催化具有一定空间位阻的酮类分子加氢的实验结果不同[12], 催化剂2在催化邻位和间位取代苯乙酮衍生物a8~a10、a12以及含有链状烷基底物苯丁酮(a13)和苯戊酮(a14)的加氢反应中, 也取得了较为优异的反应结果. 此现象与上文铼金属中心配位的PN配体个数对催化加氢性能基本没有影响的结论相吻合, 表明位阻效应对催化剂2影响有限. 此外, 上述研究结果也揭示了PN-Re(I)催化剂体系相比PNNP- Re(I), 在底物适用性方面具备一定优势.
表2 铼(I)配合物2催化酮类加氢制醇aTable 2 Catalytic hydrogenation of ketones to alcohols by 2 |
 |
a Reaction condition: 2.0 mmol ketone, 4 mL iPrOH, 0.1 mol% 2, 10 mol% NaOEt, 2 MPa H2, 100 ℃, 6 h. Isolated yield in parentheses. b 0.2 mol% 2, 20 mol% NaOEt. |
除催化苯乙酮及其衍生物加氢外, 催化剂2在催化1-茚酮(a15)、1-四氢萘酮(a16)以及脂环酮(a17~a20)等环状酮类加氢反应中也展现出一定的催化加氢活性(57%~97%). 在催化脂肪酮加氢反应中, 催化剂2对苄基丙酮(a22)、2-戊酮(a23)以及2-庚酮(a24)等的加氢催化性能更优, 目标产物收率分别达95%、69%和97%; 相较之下, 其催化得到1-环戊基乙醇(b21)的收率仅为52%. 值得一提的是, 在催化含有N、O和S等杂原子酮类分子加氢反应中, 催化剂2也展现出一定的催化加氢活性. 例如, 催化剂2催化5,6,7,8-四氢喹啉-5-酮(a26)和3-乙酰基噻吩(a29)加氢时, 产物收率分别可达90% 和64%; 在此基础上, 将催化剂2的用量增至0.2 mol%, 可有效实现4-乙酰吡啶(a25)、5-甲基-2-乙酰基呋喃(a27)以及2-乙酰基苯并呋喃(a28)的催化转化. 综上所述, PN-Re(I)催化剂体系有着良好的底物适用性.
1.4 推测的反应机理
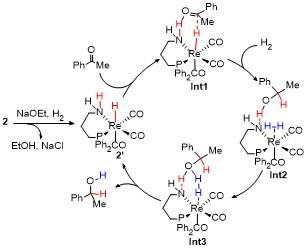
结合文献报道和本课题组前期在金属-NH配体协同催化领域的研究[15a,15b,21], 推测配合物2催化苯乙酮加氢制1-苯乙醇的反应机理. 如图5所示, 2首先在助剂NaOEt和H2共同作用下原位生成催化活性物种2', 进而通过Re-H和N-H分别与底物分子羰基C原子和O原子作用, 生成六元环中间体Int1. 紧接着, 在形成C—H键的同时H2分子与金属中心作用生成中间体Int2. 最后, 在进一步形成中间体Int3的基础上, H2分子异裂生成目标产物1-苯乙醇, 催化剂回到初始态. 与文献报道结果相似, 在整个催化反应过程中, 配体N-H仅通过非键相互作用稳定催化反应中间体[19c,19d,21]. 该机理与配合物2的加氢性能随着NaOEt的用量增加而提升的实验结果相吻合, 在一定程度上佐证了N-M′结构基元生成对提升催化活性的关键作用.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2 结论
以Re(CO)5Cl与双齿PN配体为原料, 成功制备出铼配合物1~6. 在采用核磁共振、红外以及X射线单晶衍射等技术对结构进行系统表征基础上, 将PN-Re(I)配合物应用于催化酮类分子加氢制醇, 并系统探究了配合物结构、助剂种类和用量、氢气压力以及温度等反应条件对催化性能的影响. 研究发现, 在PN-Re(I)配合物催化剂体系中, 金属中心配位的PN配体数量对催化性能无显著影响, 其中以配合物2的性能最优. 在优化反应条件下, 催化苯乙酮加氢的TOF值和TON值最高可达3500 h-1和42000. 在一定条件下, 2也可以有效催化芳香酮、脂肪酮以及杂环酮等近30多种酮类分子加氢制醇. 本研究将为进一步开发高效Re(I)-NH催化剂提供理论科学基础.
3 实验部分
3.1 仪器与试剂
甲苯、1,4-二氧六环、四氢呋喃、正己烷、异丙醇和乙酸乙酯等有机溶剂购自国药集团上海试剂公司; Re(CO)5Cl、苯乙酮等底物分子、NaOEt等购自阿拉丁或百灵威化学试剂公司. 配体Ph2P(CH2)2NH2、Ph2P- (CH2)3NH2和o-Ph2PC6H4NH2参照文献方法合成[22].
铼配合物1~6以及加氢反应产物的核磁共振测试在Bruker Advance II 400 MHz上完成, 以CDCl3或DMSO-d6为溶剂. 晶体学数据在STOE STADIVARI X-射线单晶衍射仪上采集完成. 红外光谱测试以及C、H、N元素含量测定分别在Nicolet FT-IR 330型红外光谱仪和Thermo Quest Italia SPA EA 1110型元素分析仪上完成. 配合物催化加氢活性评价采用配备有石英内衬的高压反应釜, 体积为25 mL或100 mL. 反应液采用配备有氢焰离子化检测器(FID)以及KB-Wax色谱柱(30 m×0.32 mm×0.33 µm)的气相色谱仪进行分析.
3.2 实验方法
实验中涉及的无氧无水操作均采用标准Schlenk技术或在氩气氛手套箱中进行. 有机溶剂甲苯、正己烷、四氢呋喃(THF)以及1,4-二氧六环等用钠丝预处理后, 氮气气氛下用钠钾合金回流后取用. DMSO-d6经脱气处理后, 在手套箱中用氢化钙搅拌2 d后过滤使用; CDCl3用氢化钙室温搅拌2 d后, 在氮气气氛保护下蒸馏, 置于手套箱中备用. 苯乙酮等液态底物以及异丙醇等醇类溶剂用氢化钙室温搅拌2 d后, 氮气气氛下蒸馏, 储存在手套箱中备用. NaOEt等固体药品直接经脱气处理后, 储存在手套箱中备用.
3.2.1 配合物的活性评价过程
在手套箱中准确称取或量取铼配合物、NaOEt、苯乙酮和异丙醇于反应釜内衬中, 组装好釜体并转移出手套箱. 紧接着, 用H2冲洗釜内气氛三次后充H2至2 MPa, 将釜体置于加热搅拌装置中, 按指定条件加热反应; 反应完成后, 用冰水冷却釜体至约10 ℃后排去釜中剩余的H2. 最后, 在所得反应液中加入一定量正十二烷, 搅拌均匀后用色谱检测, 或将反应液浓缩后经硅胶色谱纯化, 得到加氢反应产物. 色谱检测所用参数如下: N2为载气(0.07 MPa); 气化室和FID检测器温度分别为200和250 ℃; 程序升温过程如下: 80 ℃维持5 min, 后以10 ℃/min升高温度至200 ℃, 并维持10 min. 根据所得谱图中产物、原料以及正十二烷的峰面积进行转化率和产率的计算.
3.2.2 配合物(Ph2PCH2CH2NH2)Re(CO)3Cl (1)的合成
在氩气氛手套箱中, 称取0.18 g Re(CO)5Cl (0.5 mmol)和0.11 g配体Ph2P(CH2)2NH2 (0.5 mmol)于含有约30 mL甲苯的耐压瓶(75 mL)中, 密闭后转移出手套箱, 置于100 ℃油浴锅中搅拌加热, 反应过程中反应液由黄色逐渐变为无色透明溶液. 24 h后在惰性气氛保护下冷却反应液至室温, 减压浓缩反应液至约2 mL后加入约5 mL正己烷, 过滤收集析出的白色沉淀, 用正己烷洗涤(5 mL×3), 减压干燥得到配合物1, 0.18 g, 产率67%. 1H NMR plus 1H-13C HSQC (400 MHz, CDCl3, 298 K) δ: 2.22~2.29 (m, 1H, CH2), 2.64~2.74 (m, 1.5H, CH2), 3.00 (br, 1H, NH), 3.21~3.30 (m, 1H, NH), 3.72~3.89 (m, 1.5H, CH2), 7.40~7.50 (m, 6.5H), 7.53~7.58 (m, 2H), 7.70~7.75 (m, 1.5H) (Ph); 13C NMR (100 MHz, CDCl3, 298 K) δ: 26.55 (d, JPC=26.0 Hz, CH2), 46.00 (d, JPC=7.4 Hz, CH2), 128.37 (s), 129.13 (q, JPC=13.7 Hz), 131.09 (d, JPC=47.4 Hz), 130.90 (d, JPC=2.2 Hz), 131.09 (d, JPC=2.6 Hz), 131.54 (d, JPC=10.9 Hz), 131.88 (br), 132.29 (d, JPC=10.6 Hz), 132.33 (d, JPC=47.0 Hz), 133.07 (d, JPC=9.1 Hz), 133.57 (d, JPC=10.6 Hz) (Ph), 190.42 (d, JPC=6.5 Hz, CO), 191.77 (d, JPC=69.5 Hz, CO), 195.44 (d, JPC=6.9 Hz, CO); 31P NMR (160 MHz, CDCl3, 298 K) δ: 30.57 (s); IR (Nujol mull, KBr) ν: 2020, 1920, 1880 cm-1. HRMS (ESI) calcd for ReC17H16NPO3 [M]+ 500.0425, found 500.0429. Anal. calcd for ReC17- H16NPO3Cl: C 38.17, N 2.62, H 3.01; found C 37.38, N 2.58, H 2.98.
3.2.3 配合物(Ph2PCH2CH2CH2NH2)Re(CO)3Cl (2)的合成
在氩气氛手套箱中, 称取0.18 g Re(CO)5Cl (0.5 mmol)和0.12 g配体Ph2P(CH2)3NH2 (0.5 mmol)于含有约30 mL甲苯的耐压瓶(75 mL)中, 密闭后转移出手套箱, 置于100 ℃油浴锅中搅拌加热, 反应过程中生成白色浑浊液. 24 h后, 在惰性气氛保护下反应液冷却至室温, 过滤收集并用正己烷洗涤(5 mL×3)生成的白色固体, 减压干燥得到配合物2 0.15 g, 产率55%. 1H NMR plus 1H-13C HSQC (400 MHz, DMSO-d6, 298 K) δ: 1.82~1.88 (m, 1H, CH2), 2.00~2.11 (m, 1H, CH2), 2.59~2.66 (m, 3 H, CH2), 3.19 (d, 2JHH=8.4 Hz, 1H, CH2), 4.36 (br, 1H, NH), 4.74 (br, 1H, NH), 7.42~7.62 (m, 10H, Ph); 13C NMR (100 MHz, DMSO-d6, 298 K) δ: 22.69 (d, JPC=24.4 Hz, CH2), 23.69 (br, CH2), 40.09 (s, CH2), 128.23 (d, JPC=9.5 Hz), 128.76 (d, JPC=9.1 Hz), 130.14 (d, JPC=2.2 Hz), 130.33 (d, JPC=2.2 Hz), 132.23 (t, JPC=10.2 Hz), 132.42 (s), 132.89 (d, JPC=9.9 Hz), 133.06 (s), 133.51 (s) (Ph), 190.88 (s, CO), 191.41 (t, JPC=12.6 Hz, CO), 194.79 (d, JPC=8.4 Hz, CO); 31P NMR (160 MHz, DMSO-d6, 298 K) δ: -4.13 (s); IR (Nujol mull, KBr) ν: 2010, 1920, 1860 cm-1. HRMS (ESI) calcd for ReC18H18NPO3 514.0582, found 514.0583. Anal. calcd for ReC18H18NPO3Cl: C 39.38, N 2.55, H 3.31; found C 39.50, N 2.56, H 3.25.
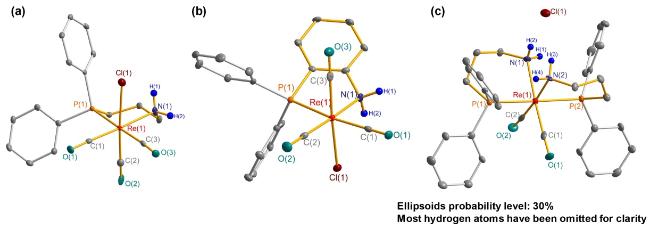
配合物2的单晶是在手套箱中, 室温下2的二氯甲烷溶液经正己烷缓慢渗透2 d长成.
3.2.4 配合物(o-Ph2PC6H4NH2)Re(CO)3Cl (3)的合成
在氩气氛手套箱中, 称取0.18 g Re(CO)5Cl (0.5 mmol)和0.14 g配体o-PPh2C6H4NH2 (0.5 mmol)于含有约30 mL甲苯的耐压瓶(75 mL)中, 密闭后转移出手套箱, 并置于100 ℃油浴锅中搅拌加热, 反应过程中生成白色浑浊液. 24 h后, 在惰性气氛保护下冷却反应液至室温, 过滤收集并用正己烷洗涤(5 mL×3)生成的白色固体, 减压干燥得到配合物3 0.15 g, 产率51%. 1H NMR plus 1H-13C HSQC (400 MHz, DMSO-d6, 298 K) δ: 2.50~2.54 (m, 1H, NH), 7.10 (dd, 2JHH=3.2, 13.6 Hz, 1H, NH), 7.27~7.32 (m, 2H), 7.40 (t, 2JHH=7.2 Hz, 1H), 7.46~7.60 (m, 9H), 7.75~7.80 (m, 2H) (C6H4, Ph); 13C NMR (100 MHz, DMSO-d6, 298 K) δ: 127.16 (d, JPC=5.3 Hz), 127.97 (s), 128.06 (d, JPC=1.7 Hz), 128.46 (d, JPC=11.19 Hz), 128.84 (d, JPC=7.6 Hz), 129.22 (s), 129.69 (s), 130.39 (d, JPC=2.5 Hz), 130.99 (d, JPC=2.8 Hz), 131.79 (d, JPC=12.1 Hz), 132.28 (s), 133.59 (s), 133.76 (s), 134.10 (s), 134.30 (d, JPC=10.4 Hz), 151.33 (d, JPC=22.6 Hz) (C6H4 and Ph), 189.92 (d, JPC=6.1 Hz, CO), 193.09 (d, JPC=71.8 Hz, CO), 196.39 (d, JPC=7.2 Hz, CO); 31P NMR (160 MHz, DMSO-d6, 298 K) δ: 28.23 (s); IR (Nujol mull, KBr) ν: 2018, 1910, 1889 cm-1. HRMS (ESI) calcd for ReC21H16NPO3 548.0425, found 548.0412. Anal. calcd for ReC21H16NPO3Cl: C 43.26, N 2.40, H 2.77; found C 43.56, N 2.29, H 2.69.
配合物3的单晶是在手套箱中, 室温下3的二氯甲烷溶液经正己烷缓慢渗透4 d长成.
3.2.5 配合物[(Ph2PCH2CH2NH2)2Re(CO)2]Cl (4)的合成
在氩气氛手套箱中, 称取0.18 g Re(CO)5Cl (0.5 mmol)和0.22 g配体Ph2P(CH2)2NH2 (1.0 mmol)于含有约30 mL 1,4-二氧六环的耐压瓶(75 mL)中, 密闭后转移出手套箱, 并置于110 ℃油浴锅中搅拌加热, 反应过程中缓慢生成白色浑浊液. 2 d后, 在惰性气氛保护下冷却反应液至室温, 过滤收集并用正己烷洗涤(5 mL×3)生成的白色固体, 减压干燥得到配合物4 0.13 g, 产率35%. 1H NMR plus 1H-13C HSQC (400 MHz, DMSO-d6, 298 K) δ: 2.39 (br, 2H, CH2), 2.74 (br, 2H, CH2), 2.90 (br, 2H, CH2), 3.05~3.09 (m, 2H, CH2), 3.95 (br, 2H, NH), 4.03 (br, 2H, NH), 7.42~7.52 (m, 12H), 7.71~7.77 (m, 8 H) (Ph); 13C NMR (100 MHz, DMSO-d6, 298 K) δ: 27.68 (t, JPC=14.4 Hz, CH2), 44.98 (t, JPC=7.2 Hz, CH2), 128.74 (t, JPC=4.9 Hz), 129.32 (t, JPC=4.6 Hz), 130.30 (d, JPC=18.0 Hz), 131.66 (dd, JPC=5.8, 5.6 Hz), 132.93 (t, JPC=20.3 Hz), 135.66 (t, JPC=25.0 Hz) (Ph), 199.93 (br, CO); 31P NMR (160 MHz, DMSO-d6, 298 K) δ: 40.20 (s); IR (Nujol mull, KBr) ν: 1912, 1815 cm-1. HRMS (ESI) calcd for ReC30H32N2P2O2 701.1497, found 701.1488. Anal. calcd for ReC30H32N2P2O2Cl: C 48.94, N 3.81, H 4.38; found C 48.99, N 3.93, H 4.45.
3.2.6 配合物[(Ph2PCH2CH2CH2NH2)2Re(CO)2]Cl (5)的合成
在氩气氛手套箱中, 称取0.18 g Re(CO)5Cl (0.5 mmol)和0.24 g配体Ph2P(CH2)3NH2 (1.0 mmol)于含有约30 mL 1,4-二氧六环的耐压瓶(75 mL)中, 密闭后转移出手套箱, 并置于110 ℃油浴锅中搅拌加热, 反应过程中生成无色透明溶液. 2 d后, 在惰性气氛保护下冷却反应液至室温, 减压浓缩反应液至约2 mL后, 加入约5 mL正己烷, 过滤收集析出的白色沉淀, 用正己烷洗涤(5 mL×3)后, 减压干燥得到配合物5 0.14 g, 产率37%. 1H NMR plus 1H-13C HSQC (400 MHz, CDCl3, 298 K) δ: 1.87 (d, 2JHH=12.4 Hz, 2H, NH), 2.07~2.19 (m, 6H, CH2), 2.36 (dd, 2JHH=11.2, 9.2 Hz, 2H, CH2), 3.12~3.21 (m, 4H, CH2), 5.68 (t, 2JHH=10.8 Hz, 2H, NH), 7.42~7.60 (m, 16H), 7.78~7.83 (m, 4H) (Ph); 13C NMR (100 MHz, CDCl3, 298 K) δ: 25.46 (s, CH2), 27.65 (t, JPC=13.3 Hz, CH2), 45.06 (s, CH2), 129.00 (t, JPC=5.1 Hz), 129.93 (t, JPC=4.0 Hz), 130.60 (s), 130.80 (s), 131.58 (t, JPC=4.7 Hz), 131.92 (t, JPC=18.7 Hz), 134.04 (t, JPC=5.8 Hz), 135.14 (t, JPC=26.6 Hz) (Ph), 200.31 (s, CO); 31P NMR (160 MHz, CDCl3, 298 K) δ: -3.25 (s); IR (Nujol mull, KBr) ν: 1910, 1810 cm-1. HRMS (ESI) calcd for ReC32- H36N2P2O2 729.1810, found 729.1804. Anal. calcd for Re- C32H36N2P2O2Cl: C 50.28, N 3.67, H 4.75; found C 50.46, N 3.51, H 4.62.
配合物5的单晶是在手套箱中, 室温下5的二氯甲烷溶液经正己烷缓慢渗透3 d长成.
3.2.7 配合物[(o-Ph2PC6H4NH2)2Re(CO)2]Cl (6)的合成
在氩气氛手套箱中, 称取0.18 g Re(CO)5Cl (0.5 mmol)和0.27 g配体o-PPh2C6H4NH2 (1.0 mmol)于含有约30 mL 1,4-二氧六环的耐压瓶(75 mL)中, 密闭后转移出手套箱并置于110 ℃油浴锅中搅拌加热, 反应过程中生成淡黄色透明溶液. 2 d后, 在惰性气氛保护下冷却反应液至室温, 减压浓缩反应液至约2 mL后加入约5 mL正己烷, 过滤收集析出的黄色沉淀, 用1,4-二氧六环重结晶得到配合物6 0.35 g, 产率84%. 1H NMR plus 1H-13C HSQC (400 MHz, CDCl3, 298 K) δ: 3.32 (d, 2JHH=13.2 Hz, 2H, NH), 8.53 (d, 2JHH=13.2 Hz, 2H, NH), 6.93 (d, 2JHH=7.6 Hz, 2H), 7.21 (t, 2JHH=7.2 Hz, 2H), 7.31 (t, 2JHH=7.6 Hz, 2H), 7.40~7.43 (m, 3 H), 7.45~7.53 (m, 11H), 7.56~7.59 (m, 4H), 7.87 (dd, 2JHH=5.6, 4.0 Hz, 4H) (28 H, C6H4 and Ph); 13C NMR (100 MHz, CDCl3, 298 K) δ: 127.03 (t, JPC=4.1 Hz), 127.61 (t, JPC=3.0 Hz), 129.09 (t, JPC=5.5 Hz), 129.74 (t, JPC=4.6 Hz), 130.58 (s), 131.41 (s), 131.68 (s), 131.91 (d, JPC=3.6 Hz), 132.11 (t, JPC=5.9 Hz), 132.20 (s), 132.28 (s), 132.47 (s), 132.58 (s), 132.81 (s), 133.06 (s), 134.50 (t, JPC=6.6 Hz), 150.82 (t, JPC=13.5 Hz) (C6H4, Ph), 200.40 (br, CO); 31P NMR (160 MHz, CDCl3, 298 K) δ: 40.65 (s); IR (Nujol mull, KBr) ν: 1930, 1840 cm-1. HRMS (ESI) calcd for ReC38- H32N2P2O2 797.1497, found 797.1493. Anal. calcd for ReC38H32N2P2O2Cl: C 54.83, N 3.37, H 3.88; found C 54.16, N 3.35, H 3.94.
辅助材料(Supporting Information) 助剂和溶剂优化活性测试结果、单晶结构数据(CCDC 2464480~2464482)以及化合物1~6和加氢反应产物的核磁共振谱图. 这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
(Lu, Y.)