

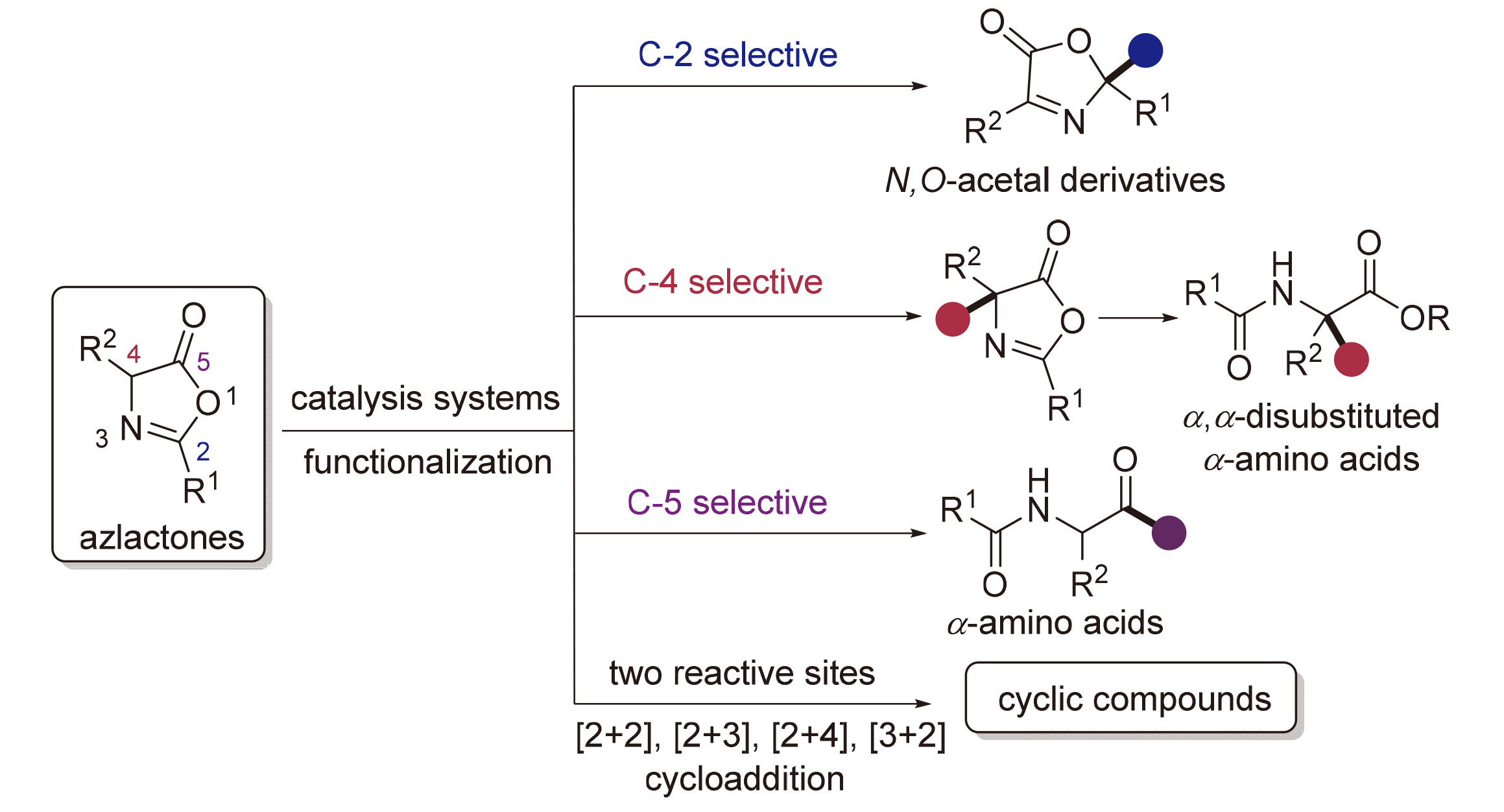
噁唑酮作为一种重要的五元杂环结构, 不仅是许多天然产物和药物分子的重要组成部分[1], 而且是合成α-季碳氨基酸和N,O-缩醛类化合物的重要起始原料[2]. 鉴于噁唑酮类化合物的重要性, 如何快速高效地构建和修饰噁唑酮类化合物, 一直是合成化学家们关注的重要研究方向.
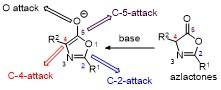
噁唑酮类化合物分子结构中同时存在多个兼具亲核性和亲电性的反应位点, 这种双亲特性使其能够参与多样化的反应路径. 噁唑酮环结构具有C-2、C-4和C-5三个反应位点, 其中C-2位和C-5位点主要表现为亲电位点, C-4位主要表现为亲核位点. 由于噁唑酮通常在碱性条件下去质子化生成烯醇中间体, 因此C-2位在一定条件下也具有亲核性, 使得噁唑酮类化合物可以参与多种不同类型的化学反应, 如C-2位或C-4位可以作为亲核取代或亲核加成的反应位点, C-2位或C-5位可以作为亲电取代或亲电加成的反应位点, 在C-4和C-5位可以发生[2+n]环加成反应, 在C-2和C-4位可以发生 [3+n]环加成反应. 此外, 利用C-5位的亲电性还可以发生开环反应(Figure 1). 本综述基于噁唑酮不同反应位点, 对近年来噁唑酮类化合物的官能团化反应进行了总结, 具体分为以下5种类型: C-2位官能团化, C-4位官能团化, C-5位开环反应, 多反应位点的环加成反应, 以及原位形成噁唑酮的官能团化.
1 C-2位官能团化
噁唑酮的亲核反应位点活性与选择性显著依赖于其取代基类型(空间位阻效应和电子效应)、手性催化剂类型及反应条件(如温度、溶剂等)的协同调控. 噁唑酮最常见的反应类型是C-4位衍生化反应, 若噁唑酮C-2位上存在强吸电子基团或者C-4位存在大位阻基团时, 加成反应发生在C-2位.
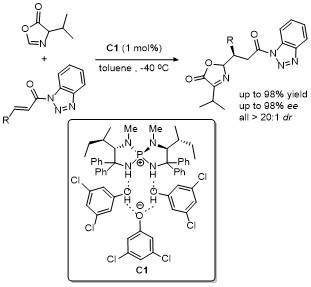
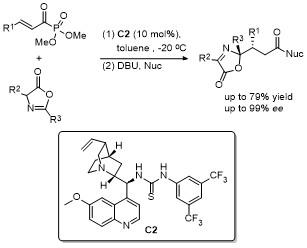
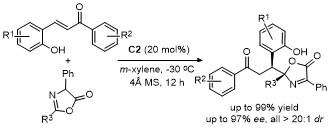
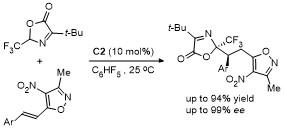
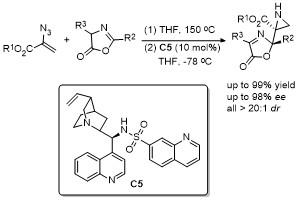
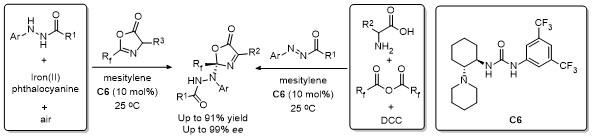
2 C-4位官能团化
2.1 C-4位烯丙基化反应
1965年, Tsuji课题组[12]报道了首例烯丙位烷基化反应. 钯催化下亲核试剂(如活性亚甲基化合物, 烯胺类化合物)对烯丙基化合物(如乙酸烯丙酯和烯丙基溴)亲核取代进行烯丙基化的反应. 目前, 不对称烯丙位烷基化(AAA)反应是构建具有对映选择性的C—C、C—N和C—O键最强大且最常用的反应之一.
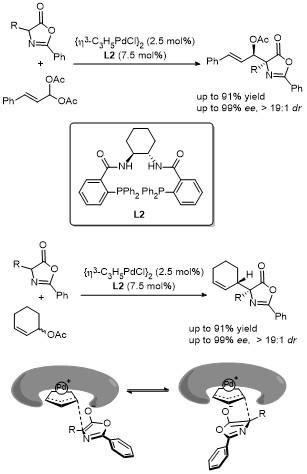
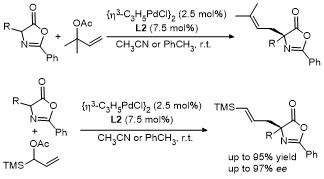
2011年, Giacomo课题组[17]利用钯(II)催化剂/双齿膦配体, 成功催化了2,3-不饱和吡喃苷与噁唑酮在C4位的加成反应. 该反应在温和条件下进行, 能够兼容多种官能团, 高效(最高90%收率)、高对映选择性(最高96% ee)地获得了一系列含有两个新立体中心的噁唑酮衍生物(Scheme 14). 该研究的关键在于反应中生成的π-烯丙基钯(II)中间体, 它与氨基酸衍生的噁唑酮发生对映选择性加成反应, 进而确保反应兼具高选择性与精准的立体控制能力. 随后, 通过二羟基化和水解反应, 最终产物可被转化为α-D-C-甘露糖醇-(S)-氨基酸. 这类兼具糖和氨基酸功能特性的化合物在药物开发和生物活性分子合成方面展现出广阔的应用前景.
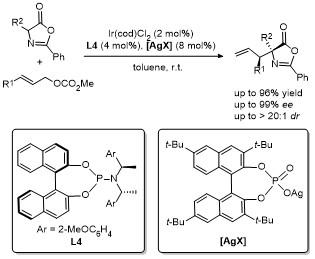
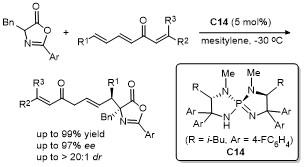
2013年, Hartwig课题组[18]报道了金属铱/亚磷酰胺配体与无光学活性磷酸阴离子的共催化体系, 实现了反式-肉桂基甲基碳酸酯与噁唑酮的不对称烯丙基化反应(Scheme 15). 该反应于室温条件下甲苯溶剂中进行, 产率高达96%, 且展现了优异的立体选择性, 非对映选择性>20∶1 dr, 对映选择性>95% ee. 该研究的关键创新点在于发现了磷酸阴离子的独特作用. 尽管金属铱与亚磷酰胺配体的络合物能够提供出色的对映选择性, 但对于非对映选择性的控制效果有限. 加入无光学活性的磷酸阴离子后, 非对映选择性显著提高, 成功控制了反应的区域选择性和立体选择性. 进一步实验揭示了磷酸阴离子的结构多样性对非对映选择性的显著影响, 通过精细调控不同类型的磷酸阴离子, 可以进一步优化反应选择性. 这项研究为不对称烯丙化反应提供了全新途径, 展示了金属铱与亚磷酰胺配体的巨大潜力, 并为高效且高选择性的化学反应设计提供了新的思路.
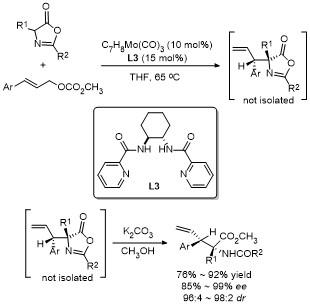
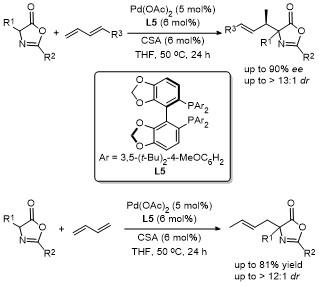
2018年, 邢栋课题组[19]报道了无碱条件下钯催化噁唑酮与1,3-二烯的高度非映对和对映选择性烯丙基烷基化反应(Scheme 16). 该反应由Pd(OAc)2/SEGPHOS和樟脑磺酸组成的催化体系, 以取代的1,3-二烯为原料,经1,2-加成反应合成了一系列具有良好非对映选择性和高立体选择性的噁唑酮类化合物. 值得注意的是, 以简单的1,3-丁二烯为烯丙基前体, 得到了收率高、区域选择性高的1,4-加成产物. 这项研究提供了一种高效的无碱条件下钯催化不对称烯丙基烷基化方法, 拓展了1,3-二烯类化合物的反应范围, 同时展示了在合成具有高立体选择性复杂分子中的巨大潜力, 进一步推动了不对称催化领域的发展.
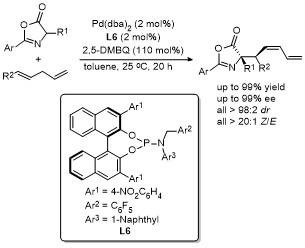
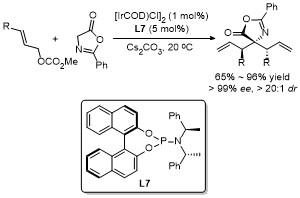
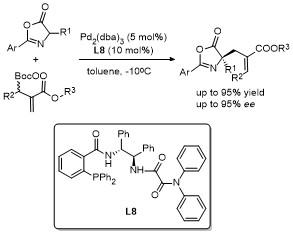
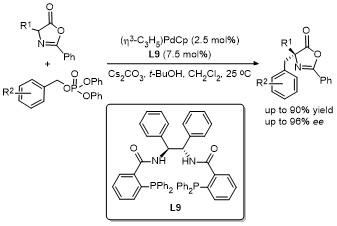
2.2 C-4位烷基化反应
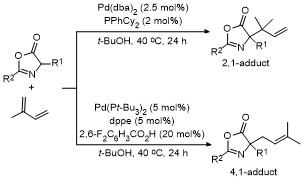
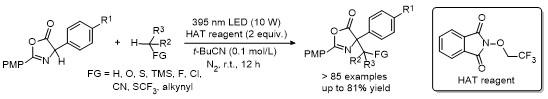
2.3 C-4位炔基化反应
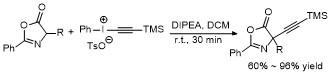
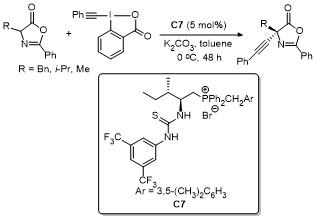
2.4 C-4位芳基化反应
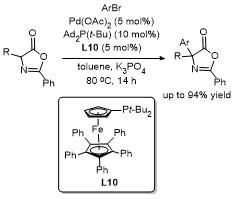
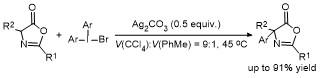
2.5 C-4位Steglich重排
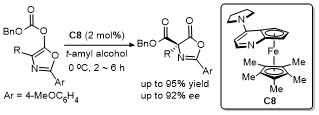
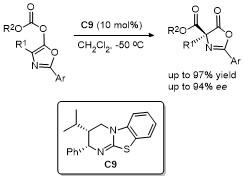
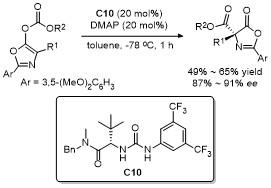
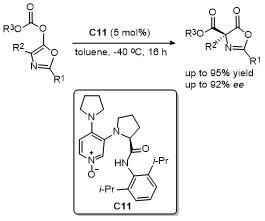
2.6 C-4位Michael加成反应
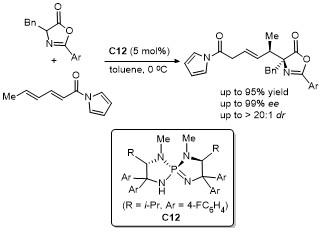
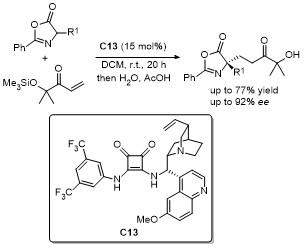
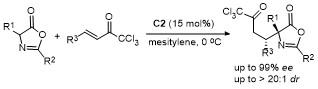
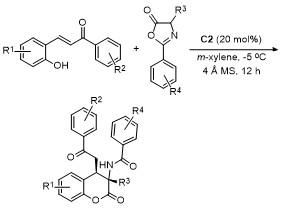
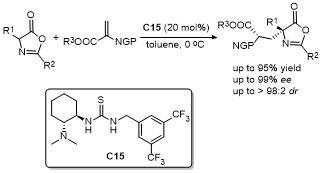
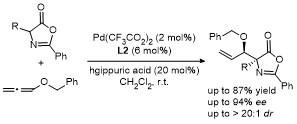
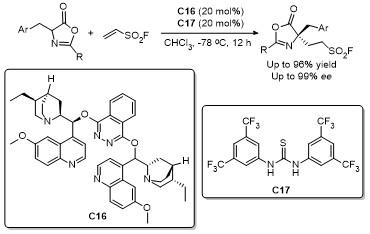
2021年, 鄢明课题组[42]报道了一项创新研究, 通过(DHQD)₂PHAL和硫脲的协同催化实现了噁唑酮与乙烯磺酰氟的对映选择性共轭加成反应, 合成了2-苯基-4-苄基噁唑酮(Scheme 38). 该反应不仅展现了极高的收率(最高可达96%), 还具有卓越的对映选择性(99% ee), 为含磺酰氟基团的手性噁唑酮的合成提供了一种便捷且高效的途径. 此外, 反应产物可通过简单的后续操作转化为具有药用价值的手性磺内酰胺和α-季氨基酸衍生物, 这类化合物在药物研发中具有重要应用潜力. 该研究拓展了噁唑酮化合物的合成策略, 同时为手性磺酰氟类分子的合成提供了新工具, 展现了在药物化学和不对称合成领域的广泛应用前景.
2.7 C-4位Mannich反应
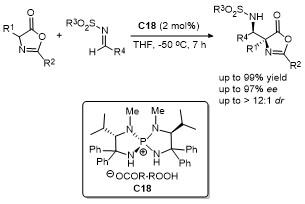
2012年, 龚流柱课题组[44]报道了一种新型手性双碱催化剂, 成功应用于噁唑酮与脂肪族亚胺的Mannich反应(Scheme 40). 这种催化剂凭借其独特的手性甜菜碱结构设计, 展现了卓越的催化效果. 研究表明, 这些有机碱基通过多功能协同作用, 显著促进了噁唑酮与多种脂肪族亚胺的反应, 生成的产物具有极高的对映选择性(94%~99% ee). 此外, 研究还揭示了该催化剂的可调节性. 通过调整催化剂中的联萘基和金鸡纳生物碱序列, 催化剂结构可以根据不同反应需求灵活改变, 从而适应不同的底物和反应条件. 这种灵活性使得该催化体系不仅适用于优化现有的Mannich反应, 还为开发其他不对称催化反应提供了新思路.
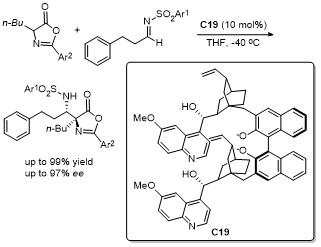
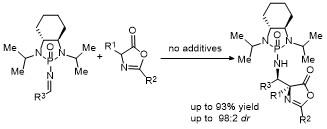
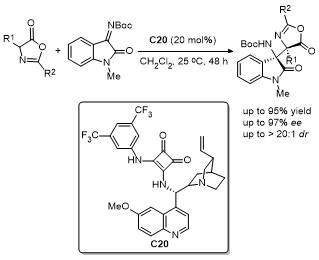
2.8 C-4位Ene-type反应
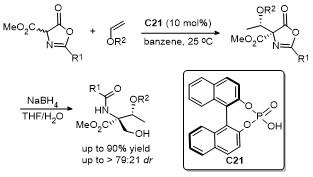
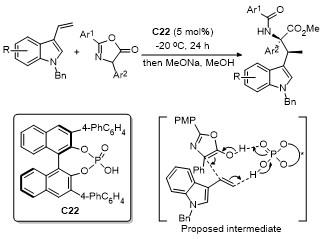
2.9 C-4位Aldol反应
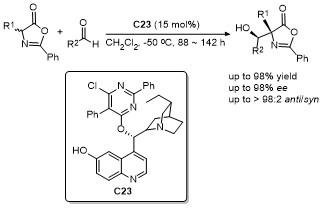
2.10 C-4位其它衍生化反应
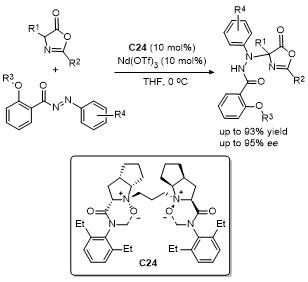
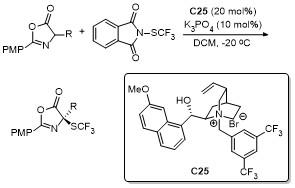
3 C-5位不对称动态动力学拆分开环反应
动态动力学拆分(DKR)是手性合成领域高效制备光学纯多手性化合物的理想方法, 可将外消旋底物的两种对映体转化为单一对映体产物, 这种巧妙的策略可以克服传统“动力学拆分”最大理论产率50%的限制. 因此, DKR在构建具有多个连续立体中心的手性分子方面展示了巨大的潜力, 尤其是在手性药物和手性催化剂的生产中具有重要应用价值. 噁唑酮的C-5位具有亲电性, 可以发生动态动力学拆分开环反应. 随着近年来科研工作者对于噁唑酮类化合物的不断探索, 通过选用合适的手性催化体系, 借助消旋底物一对对映异构体与手性催化剂之间存在的“匹配-不匹配”效应, 最终诱导生成具有高对映选择性和高非对映选择性产物.
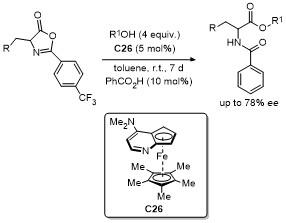
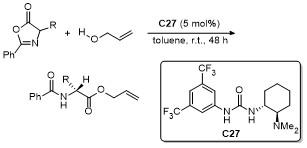
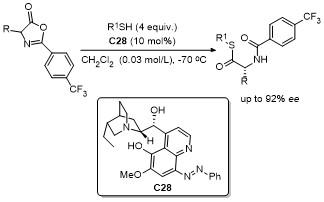
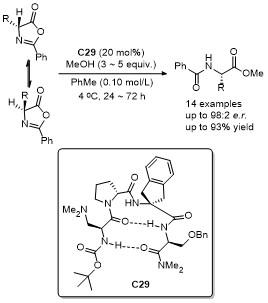
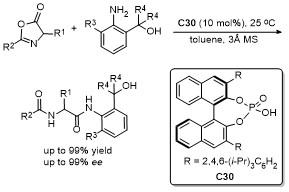
4 噁唑酮多反应位点参与的环加成反应
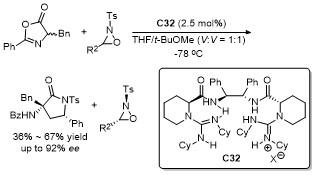
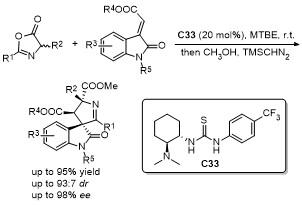
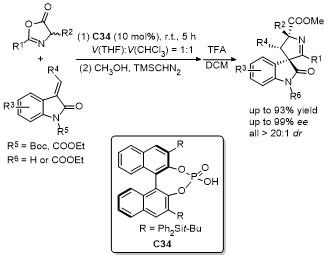
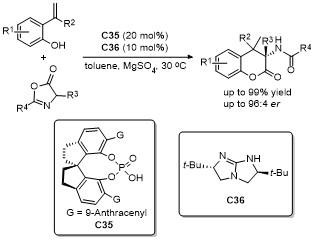
噁唑酮作为一种含有多个反应位点的五元环化合物, 其结构中的C-2、C-4、C-5具有独特的化学反应活性, 使其能够参与多种环加成反应. 这些反应在合成复杂的多环结构、构建手性中心以及设计具有生物活性的分子中具有重要的应用价值. 近年来, 对于噁唑酮环加成反应的研究报道不断涌现.
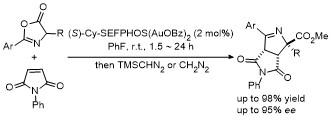
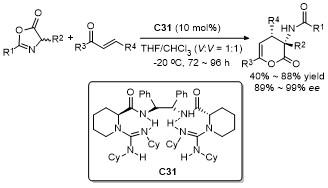
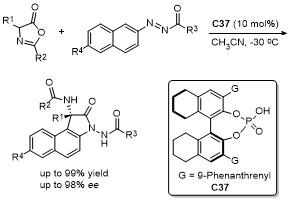
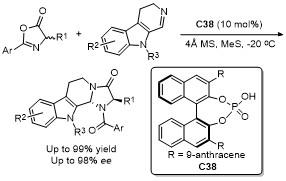
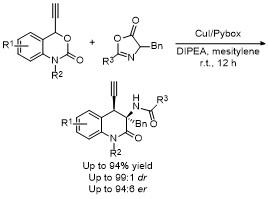
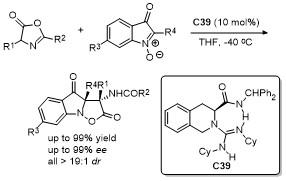
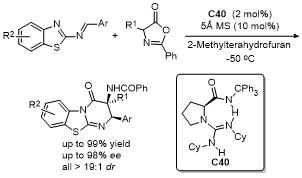
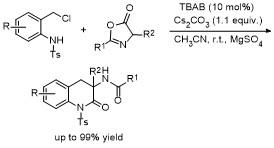
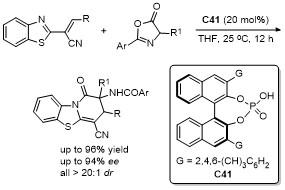
5 原位形成噁唑酮中间体的官能团化
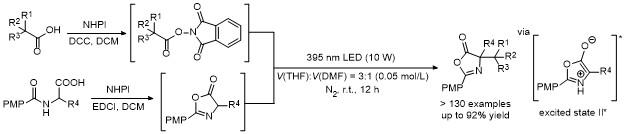
一直以来, 基于噁唑酮不同活性位点的官能团化反应层出不穷, 但噁唑酮底物的合成步骤繁琐且不稳定, 尤其是极其不稳定的2,4-二烷基取代噁唑酮, 目前还没有合适的方法合成. 因此, 基于该类噁唑酮底物的官能团化也无法实现, 而相对于研究较多的2-芳基-4-烷基噁唑酮, 2-烷基-4-芳基和2,4-二芳基噁唑酮底物的官能团化也报道较少.
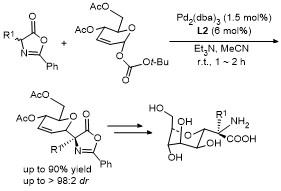
基于此, 最近我们课题组[70]开发了一种高效快速原位形成噁唑酮的策略, 利用α-氨基酸衍生的N-羟基邻苯二甲酰羟胺酯(NHP酯), 在碱作用下发生分子内环化脱去一分子邻苯二甲酰羟胺, 同时生成噁唑酮中间体. 随后, 该噁唑酮中间体发生各种官能团转化, 例如在过渡金属铜催化下, 能与炔丙基酯发生C-4位不对称炔丙基化反应, 以高收率和高立体选择性得到一系列重要的手性α-炔丙基取代的α-氨基酸衍生物(Scheme 66); 而在过渡金属钯催化下, 能与常用的烯丙基底物发生不对称烯丙基化反应, 合成各种光学纯的α-烯丙基取代的α-氨基酸衍生物(Scheme 66)[71]. 该策略避免了合成不稳定的噁唑酮, 通过原位生成的策略实现各种取代噁唑酮底物的官能团化. 值得一提的是, 该策略成功实现了2,4-二烷基取代噁唑酮底物的不对称转化, 弥补了已有噁唑酮官能团化反应的不足.
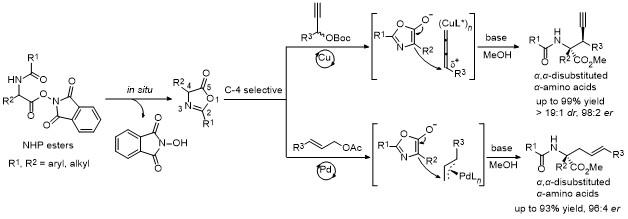
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
6 结论与展望
噁唑酮类化合物作为五元杂环化合物, 凭借其C-2、C-4和C-5位点的独特反应活性, 在有机合成、药物开发及材料科学等领域展现出重要价值. 近年来, 针对不同反应位点的官能团化及衍生化研究取得显著进展, 为高效构建复杂手性分子和生物活性化合物提供了多样化策略. 当前研究通过新型催化剂设计(如手性磷酸、双功能有机催化剂)及反应机制创新, 显著提升了噁唑酮衍生物的合成效率和立体选择性.
展望未来, 噁唑酮化学的发展应重点关注以下方向: (1)开发新型稳定化策略, 如引入保护基团或设计空间位阻结构, 以解决噁唑酮底物不稳定的关键问题; (2)发展模块化合成方法, 通过一锅多步反应简化合成步骤, 提高原子经济性; (3)深化原位生成噁唑酮策略研究, 重点开发光催化、电催化等绿色催化体系, 实现传统方法难以合成的敏感底物转化; (4)拓展不对称催化应用, 设计新型手性配体以实现高对映选择性转化; (5)加强计算化学辅助的机理研究, 为新型反应设计提供理论指导. 这些策略的实施将显著提升噁唑酮在抗肿瘤药物、抗生素等生物活性分子合成中的应用价值, 并为功能材料的分子设计开辟新途径.
(Cheng, F.)