


Nitrogen-containing heterocycles are ubiquitous molecular architectures characterized by the substitution of nitrogen atoms at one or more skeletal positions. These structures exhibit diverse bioactivities, including antibacterial,[1] anticancer,[2] and antiviral,[3] and it is widely found in a variety of drug structures. Their versatility also extends to agrochemical development.[4-5] The wide range of applications of these compounds has greatly piqued the interest of organic chemists in their synthesis.
The advent of green chemistry has transformed synthetic methodologies by prioritizing the systematic redesign of chemical processes. Central to this approach is the elimination of hazardous substances and the reduction of environmental impact through atom-efficient reactions. This framework promotes eco-friendly synthetic pathways that avoid toxic reagents, hazardous solvents, and harmful byproducts, while emphasizing energy optimization. In parallel, photoelectrochemical synthesis has emerged as a sustainable platform for chemical synthesis. These systems enable regioselective synthesis of nitrogen-containing heterocycles under mild reaction conditions, addressing longstanding challenges in traditional methods.
Photocatalytic strategies drive reactions by generating highly reactive species through the excitation of catalysts by light: after semiconductor materials (such as TiO2, g-C3N4) absorb photons, electrons transition from the valence band to the conduction band to form electron-hole pairs, which separate and migrate to the surface to participate in reduction and oxidation reactions respectively. Non-semi-condu- ctor materials (such as MOFs, organic dyes) rely on molecular orbital excitation (HOMO→LUMO), and activate substrates through electron transfer (such as electron injection from sensitizers to acceptors) or energy transfer. Both types of systems utilize photogenerated charges (electrons/ holes or excited states) to convert light energy into chemical energy, achieving the activation of small molecules (such as water splitting, CO2 reduction) or organic synthesis. These advancements are in line with the principles of green chemi- stry, demonstrating how innovative technologies can strike a balance between synthetic efficiency and environmental sustainability. Notably, the recent breakthrough advances in skeletal editing strategies (such as photomediated ring contraction, single-atom nitrogen insertion, or pyridine-to- pyrazole conversion) enable the precise single-atom modification of molecular frameworks. These methodologies achieve highly efficient and selective structural remodeling in complex natural product synthesis. Crucially, they circumvent the need for toxic reagents and strategically leverage light energy to suppress byproduct formation. This intrinsically embodies the principles of green chemistry: atomic economy and environmental sustainability.[6] Based on this, this article reviews eight relatively advanced methods for constructing nitrogen-containing heterocycles under photocatalytic drive in the past five years.
1 Light-driven catalytic construction of benzimidazole and its derivatives
Benzimidazoles (BIs) are heterocyclic compounds containing a benzene ring and two nitrogen atoms. Drugs featuring a benzimidazole nucleus possess unique structural characteristics and electron-rich environments that facilitate their binding to various physiologically significant sites, resulting in a range of pharmacological effects.[7] Consequently, benzimidazoles are frequently incorporated into the structures of drugs, such as omeprazole (an anti-ulcer medication) (Figure 1), flubendazole (an anti-parasitic agent), and bilastine (an antihistamine). Additionally, benzimidazoles serve as important intermediates in drug synthesis, transition metal ligands for organic reactions, and applications in material science.[8-9] The synthesis of benzimidazoles and their derivatives is particularly appealing due to the promising potential of their applications.
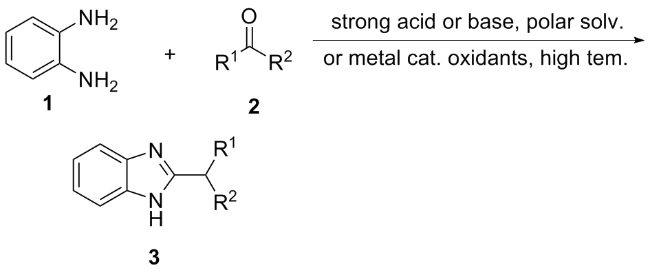
The most established conventional method for synthesizing benzimidazole is the Philips-Sladenburg synthesis (Scheme 1). In this method, benzimidazole 3 is synthesized by condensing benzenediamine and its derivatives 1 with aldehyde 2 in the presence of oxygen, followed by heating and refluxing at 100 ℃. A significant disadvantage of this method is its reliance on unstable aldehydes as feedstock, along with an excess of strong oxidizing agents such as 2,3-dichloro-5,6- dicyanobenzoquinone (DDQ)[10] and potassium persulfate (OXONE).[11] Many of these strong oxidizing agents are environmentally harmful and challenging to remove from the final product. Consequently, there is a pressing need for new synthetic methods for benzimidazole that are milder, more environmentally friendly, and suitable for industrial applications. In recent years, numerous methods utilizing light driven catalytic synthesis of benzimidazole and its derivatives have emerged, demonstrating both green and efficient characteristics.
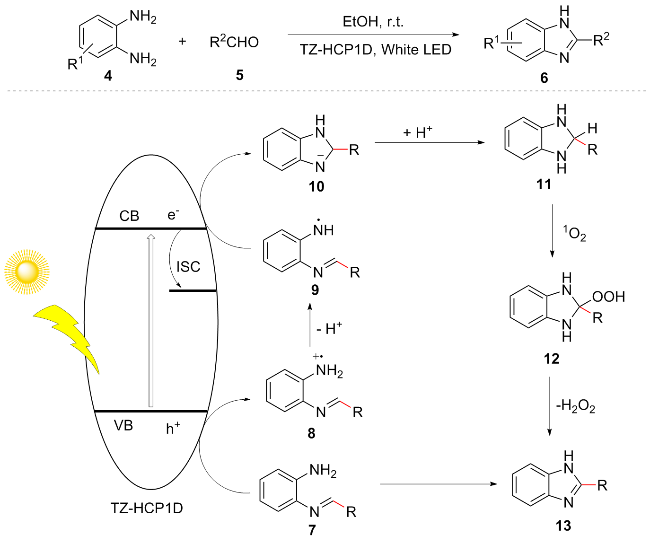
Most organic molecules cannot absorb visible light, necessitating the use of a catalyst for the photocatalytic synthesis of compounds. In 2021, An et al.[12] constructed two hyper crosslinked polymers (TZ-HCP1D and TZ-HCP2) containing s-triazine units through Friedel-Crafts alkylation reaction. Benzimidazole compound 6 was synthesized under light irradiation using o-phenylenediamine 4 and aldehyde 5 as substrates. The reaction mechanism proceeds as follows: photoexcitation generates a hole (h+) that oxidizes imine 7 to compound 8, which subsequently loses a proton to yield the free radical 9. Following cyclization, radical 9 is reduced by an electron (e) to form intermediate 10. Concurrently, the excited-state electron undergoes intersystem crossing (ISC) to produce singlet oxygen (1O2), while intermediate 10 is protonated to afford compound 11. This species then reacts with 1O2 to generate intermediate 12. Finally, 12 eliminates H2O2 to yield the final product 13 (Scheme 2). This method exhibits excellent performance in terms of cycle stability, with catalytic activity remaining above 96% after 21 cycles, as well as substrate versatility, demonstrating compatibility with aromatic aldehydes, heterocyclic aldehydes, aliphatic aldehydes, and bulky substrates.
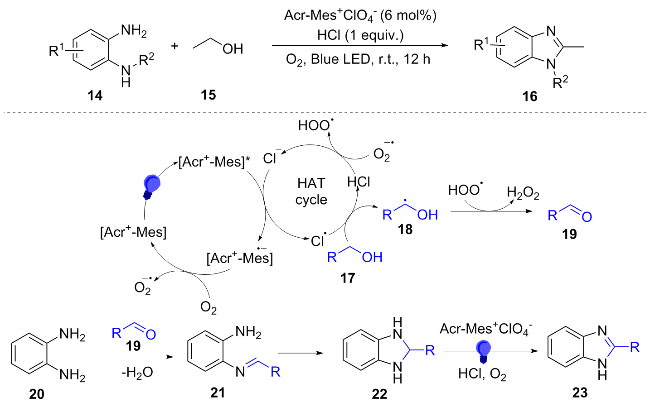
In 2023, Kumari et al.[13] synthesized benzimidazole derivatives 16 by the reaction of ethanol 15 with o-phenylene-diamine compounds 14 through the synergistic effect of a photocatalyst (Acr-Mes+ClO4) and a HAT reagent (HCl). Under blue light excitation, the photocatalyst (Acr-Mes+) is quenched through Cl reduction to produce Cl·, while the reduced form of the catalyst donates electrons to O2, generating $\text{O}{{_{2}^{}}^{-\ \bullet }}$. The Cl• radical abstracts the α-hydrogen from alcohol 17 to yield carbon radical 18, which is subsequently oxidized by $\text{O}{{_{2}^{}}^{-\ \bullet }}$ to form aldehyde 19, with concomitant release of H2O2. Aldehyde 19 then condenses with o-phenylenediamine 20 to afford benzimidazole 23 (Scheme 3). This methodology eliminates the need for transition metals, high temperatures, or strong alkaline conditions typically required in conventional approaches, enabling an efficient transformation of biomass-derived ethanol into high-value chemicals. Moreover, the reaction has been successfully scaled up to the gram level.
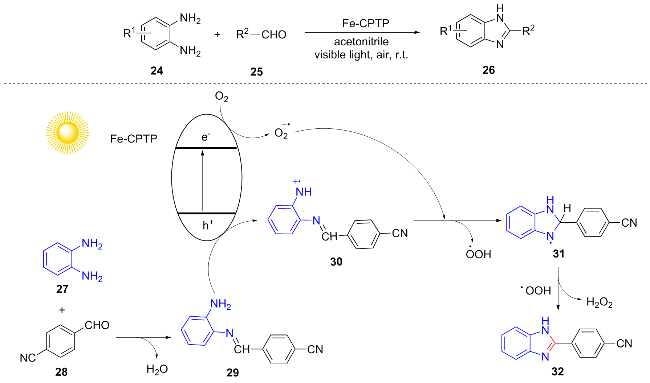
Supramolecular assembly strategies can significantly enhance the photocatalytic performance of metal complexes. Applying this strategy to the synthesis of photocatalysts can provide new ideas for green synthesis. In 2024, Zhao et al.[14] constructed a supramolecular assembly based on iron-tripyridine complex (Fe-CPTP), which efficiently catalyzed the Phillips-Ladenburg reaction of o-phenylene-diamine 24 with aromatic aldehydes 25 under room temperature, air atmosphere and visible light irradiation to synthesize 2-substituted benzimidazole compounds 26. Condensation of o-phenylenediamine 27 with 4-cyano-benzaldehyde 28 yields imine 29. Upon photoexcitation, Fe-CPTP generates the excited state species Fe-CPTP*, which transfers electrons to O2 to produce $\text{O}{{_{2}^{}}^{-\ \bullet }}$ and is concurrently oxidized to Fe-CPTP+. Fe-CPTP+ abstracts an electron from imine 29, regenerating the catalyst and simultaneously forming the imine radical cation 30. Radical cation 30 undergoes oxidative cyclization and dehydrogenation, promoted by $\text{O}{{_{2}^{}}^{-\ \bullet }}$, to yield radical cation 31. Subsequently, HOO• abstracts a proton from 31, resulting in the formation of H2O2 and the release of benzimidazole product 32 (Scheme 4). This catalytic system demonstrates excellent performance, maintaining consistent activity over 20 cycles with no detectable leaching of metal ions.
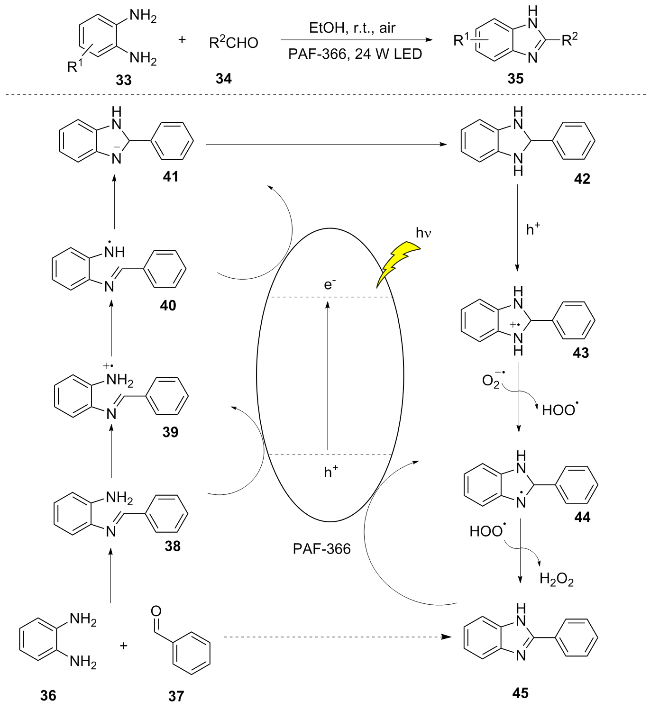
In recent years, significant advancements have been made in the field of photocatalytic energy conversion utilizing donor-acceptor (D-A) type photocatalysts. Porous aromatic frameworks (PAFs), which are visible light-active and reusable, present a green and sustainable alternative to traditional metal-based photocatalysts. In 2024, Wang et al.[15] developed three novel tetraphenylethylene (TPE)- based porous aromatic frameworks (PAFs) photocatalysts for the efficient synthesis of benzimidazole 35 compounds under visible light. Visible light excitation of PAF-366 generates electron-hole pairs (e/h+). The condensation of o-phenylenediamine 36 with benzaldehyde 37 yields imine 38, which is oxidized by h+ to form intermediate 39. Subsequent deprotonation leads to compound 40, which undergoes cyclization and is reduced by electrons (e) to afford intermediate 41. Further deprotonation produces species 42, which is oxidized by h+ to generate the nitrogen- centered radical cation 43. Through hydrogen abstraction and deprotonation mediated by $\text{O}{{_{2}^{}}^{-\ \bullet }}$, a hydrogen atom transfer process ultimately yields 2-phenylbenzimidazole 45 (Scheme 5). This work, through the rational design of D-A type PAFs, achieved efficient and green synthesis of benzimidazoles, providing new ideas for the development of porous organic photocatalysts.
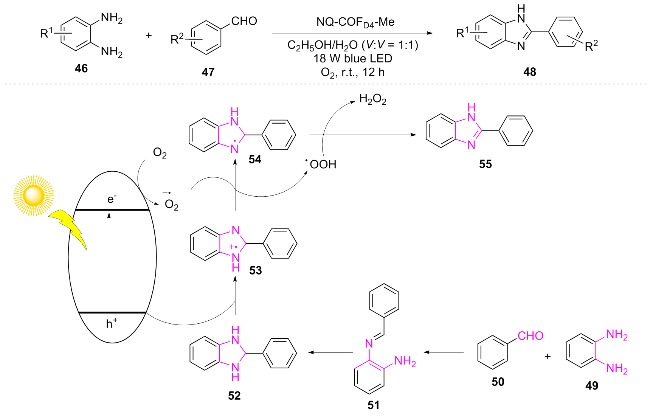
In the same year, Huang et al.[16] proposed a method to reduce the exciton binding energy of covalent organic frameworks (COFs) through ionization strategy for the efficient photocatalytic synthesis of benzimidazole 48. They proposed a reasonable mechanism: substrate 49 and 50 react via Schiff base to form imine intermediate 51, which then undergoes intramolecular cyclization to yield 52. Blue light excitation of the catalyst generates photogenerated holes (h+) and electrons (e). 52 is oxidized by h+ to form radical cation 53, while e reduces O2 to generate $\text{O}{{_{2}^{}}^{-\ \bullet }}$. $\text{O}{{_{2}^{}}^{-\ \bullet }}$ promotes the deprotonation of 53 to form radical 54 and •OOH, which couple to produce product 55 (Scheme 6). This method demonstrated extremely high product yields (91%~98%) and excellent recyclability (≥5 times) in the synthesis of benzimidazole, showcasing the great application potential of ionic COFs in photocatalytic organic synthesis.
In addition, Li et al.[17] developed WO3-ZnO composite nanomaterials in the same year, which possess dual functions of anti-gastric cancer activity and photocatalytic synthesis of benzimidazole derivatives. Under green light irradiation, it efficiently catalyzes the condensation of aromatic aldehyde 57 with o-phenylenediamine 56 to synthesize 2-substituted benzimidazole compounds 58 (Scheme 7), with yields ranging from 87% to 97%. This work innovatively applied WO3-ZnO to photocatalytic drug synthesis and anti-cancer research. The catalyst can be reused five times while maintaining high activity (yield decrease <5%), but further validation of in vivo efficacy and simplification of the preparation process are needed.
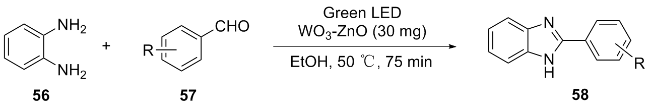
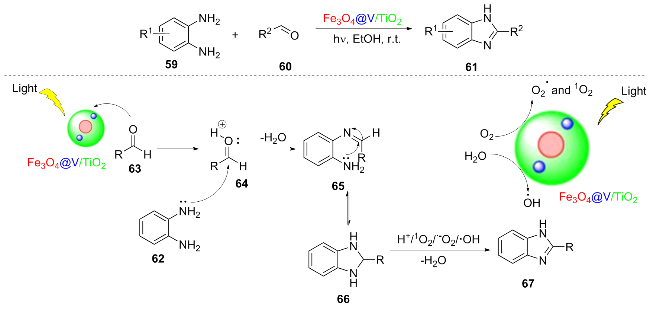
The strategy of applying nanomaterials to photocatalysts has gained widespread attention. In 2025, Bao et al.[18] developed a magnetic and recyclable Fe3O4@TiO2 nano- hetero-geneous photocatalyst for the efficient synthesis of 2-substituted benzimidazole compounds 61 from o-phenyl- enediamine 59 and aromatic aldehydes 60 under visible light. The reasonable mechanism of this reaction is as follows: Fe3O4@V/TiO2 is activated by light to generate oxonium ion 64 from aldehyde 63. The amino group of o-phenylenediamine 62 then attacks the carbonyl carbon, dehydrating to form a mono-Schiff base 65, which further undergoes intramolecular addition to yield a benzimidazoline intermediate 66. Meanwhile, the catalyst generates h+ under light and catalyzes the conversion of H2O/O2 into reactive oxygen species, such as 1O2, $\text{O}{{_{2}^{}}^{-\ \bullet }}$, and •OH, ultimately oxidizing the intermediate to 2-substituted benzimidazole 67 (Scheme 8). After seven cycles of reuse, the catalyst retains more than 80% its initial activity, offering a novel approach for the green synthesis of pharmaceutical intermediates.
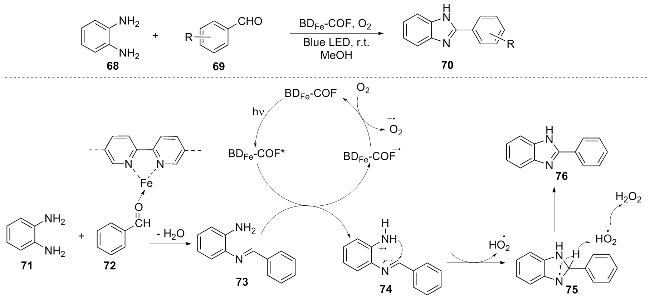
In 2025, Li et al.[19] synthesized a novel bipyrrole-Fe(III)-based covalent organic framework (BDFe-COF) through the metallization reaction of FeCl3•6H2O with the bipyrrole- based BD-COF. This organic framework can act as a photocatalyst to synthesize benzimidazole compounds 70 under light irradiation. The author proposed a reasonable mechanism: the Lewis acid sites of BDFe-COF activated benzal-dehyde 72, which condensed with o-phenylene-diamine 71 to form an imine intermediate 73. The photoexcited catalyst was single-electron reduced by this intermediate, while oxygen oxidized the catalyst to regenerate and produce $\text{O}{{_{2}^{}}^{-\ \bullet }}$, which then abstracted a proton from the intermediate to trigger intramolecular cyclization. Finally, through hydrogen transfer and dehydrogenation, the benzimidazole product 76 was generated (Scheme 9). This work achieved the efficient photocatalytic synthesis of benzimidazole compounds through the precise design of Fe3+-modified COF materials, demonstrating potential applications in green chemistry and heterogeneous catalysis.
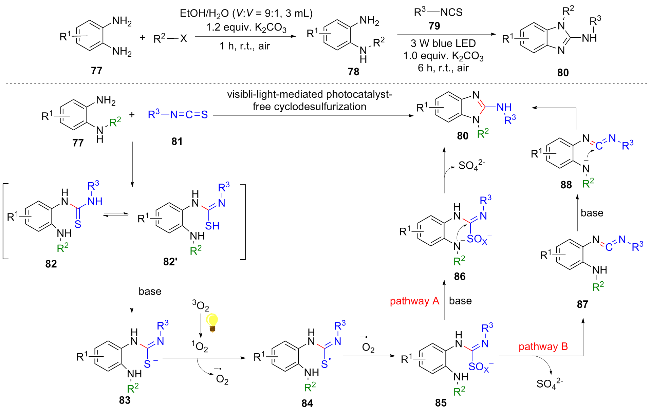
In recent years, research has been conducted on photocatalytic reactions without the need for a photocatalyst. In 2025, Rerkrachaneekorn et al.[20] developed a novel one-pot method for the synthesis of N-substituted 2-aminobenzimi-dazoles under visible light without a photocatalyst. The condensation of o-phenylenediamine 77 with isothiocyanate 81 yields thiourea 82, which undergoes deprotonation to generate the thiolate anion 83. Visible light irradiation leads to the generation of singlet oxygen (1O2), which reacts with 83 to produce a sulfur-centered radical 84 and a superoxide radical. The coupling of sulfur radical 84 with the superoxide radical forms a peroxysulfur intermediate 85, which follows two distinct reaction pathways: in Path A, 85 undergoes deprotonation and direct cyclization to yield the final product 80, accompanied by the release of sulfate ion ($\text{SO}_{\text{4}}^{2}$); in Path B, 85 first releases $\text{SO}_{\text{4}}^{2}$ to generate carbodiimide 87, which subsequently deprotonates and cyclizes to afford product 80 (Scheme 10). This method has advantages such as no need for metal catalysts and simple operation. Notably, in gram-scale experiments, the product yield can reach 85%, further verifying its potential for industrial application.
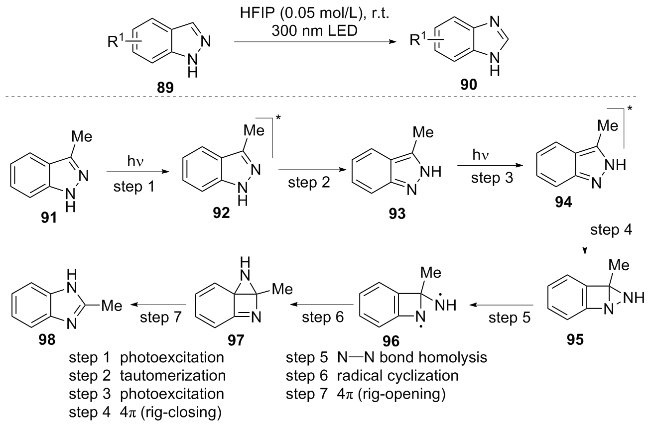
In the same year, Dos Santos et al.[21] reported a novel photocatalytic strategy for the direct conversion of indazoles 89 to benzimidazoles 90. This approach leverages photoexcitation under 300 nm UV irradiation in hexafluoroisopropanol (HFIP) solvent: 1H-Indazole first undergoes phototautomerization to its 2H-isomer, which upon further photoexcitation triggers molecular backbone rearrangement to afford the benzimidazole product (Scheme 11). The reaction features operational simplicity, high efficiency, excellent yields, and broad functional group tolerance (compatible with alkyl, halogen, alkene substituents, and applicable to chiral molecule synthesis). Its primary limitation is a narrow substrate scope.
In 2025, Wang et al.[22] has also developed a novel photocatalytic method for synthesizing benzimidazoles. This approach involves blue-light irradiation of indole and N- nitrosomorpholine, initiating ring-opening to form a key oxime intermediate. Subsequent Beckmann rearrangement completes benzimidazole ring construction. Its primary advantage is enabling direct late-stage functionalization of drug molecules (e.g. naproxen and febuxostat), effectively achieving "molecular surgery." The methodology also facilitates oxidation-mediated synthesis of fused bicyclic benzimidazoles (Scheme 12). A key limitation is its sensitivity to electron-withdrawing substituents, and phenyl substitution can lead to indazole byproduct formation. This strategy can be extended to synthesize benzoisoxazoles/ benzooxazoles (from indazole) and benzofurans, providing efficient access to diverse N-heterocyclic scaffolds.
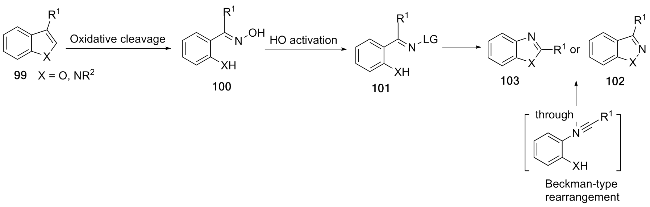
In 2025, Bartholomew et al.[23] reported an innovative photocatalytic strategy for synthesizing benzimidazoles via a light-induced nitrogen-to-carbon rearrangement of the indazole scaffold. This method utilizes N2-alkylated indazoles (e.g., methyl, benzyl derivatives) as key substrates (Scheme 13). The reaction proceeds efficiently under irradiation with UVB light (310 nm) in dimethoxyethane (DME) solvent, without requiring rigorous deoxygenation or anhydrous conditions. A significant advantage is the direct construction of asymmetric N-substituted benzimidazoles in up to 98% yields, a challenging feat using traditional alkylation methods. Furthermore, the process is scalable via continuous flow chemistry, achieving gram- scale production with 76% yield. However, substrate scope limitations exist: reactivity is restricted to N2-alkyl groups (aryl/vinyl substituents are ineffective), and electron- withdrawing groups at the C5 position can trigger competitive side reactions. This transformation exploits the red- shifted absorption characteristics of the 2H-indazole isomer, providing a novel approach for the precise synthesis of benzimidazoles.

Compared with traditional synthesis methods that rely on strong oxidants, high temperatures, and unstable aldehydes, photocatalytic synthesis of benzimidazole offers notable advantages: mild reaction conditions (room temperature and visible/UV light irradiation), elimination of toxic oxidants, and alignment with green chemistry. Different photocatalytic systems exhibit distinct characteristics. First, in terms of substrate applicability and limitations, polymer-based catalysts (e.g., TZ-HCP, PAF-366) and metal complexes (e.g., Fe-CPTP) show broad compatibility with various aldehydes, though reactivity decreases with o-phenylene- diamines bearing strong electron-withdrawing groups or sterically hindered aldehydes. Dos Santos et al.ʼs[21] UV- driven method (2025) directly converts indazoles to benzimidazoles via phototautomerization in HFIP, offering high efficiency and tolerance for alkyl/halogen/alkene groups, but suffers from a narrow substrate scope. Catalyst-free systems (e.g., Rerkrachaneekorn[20]) are specific to N-substituted 2-aminobenzimidazoles and tolerate isothiocyanates, though phenolic hydroxyl groups require protection. Wang et al.ʼs[22] blue-light method (2025) employs indole and N-nitrosomorpholine, enabling late-stage functionalization of drug molecules (e.g., naproxen) via oxime intermediates and Beckmann rearrangement, but is sensitive to electron-withdrawing groups and may form indazole byproducts with phenyl substitution. It also extends to fused bicyclic benzimidazoles and other N-heterocycles. Furthermore, regarding industrial potential, the Acr-Mes+/HCl system[13] (2023) and catalyst-free system[20] (2025) both achieve >85% yield in gram-scale synthesis. Finally, me- chanistically, most systems use ROS-mediated (ROS: Reactive oxygen species) pathways, while Fe-CPTP and BDFe- COF utilize metal-center activation. The methods developed by Dos Santos and Wang exhibit unique photochemical rearrangement pathways (phototautomerization and Beckmann rearrangement, respectively). Bartholomew et al.ʼs[23] UVB-driven strategy (2025) provides an innovative route via light-induced nitrogen-to-carbon rearrangement of N2-alkylated indazoles in DME solvent, achieving high yields (up to 98%) of asymmetric N-substituted benzimidazoles under non-rigorous conditions. However, this method is strictly limited to N2-alkyl substituents.
Photocatalytic methods, utilizing light-driven rearrange- ments, heterogeneous materials, or cooperative catalysis, enable efficient and green benzimidazole synthesis. The gram-scale amplification of alcohol activation pathways, catalyst-free systems, and innovative drug functionalization strategies highlight promising industrial potential. Future research should focus on enhancing photoresponsive material performance and broadening substrate compatibility to overcome limitations like narrow scope or substituent sensitivity, facilitating practical industrial implementation.
2 Light-driven catalytic construction of indole and its derivatives
Indole is a compound consisting of a pyrrole and benzene structure, and its derivatives are nitrogenous heterocycles found in numerous natural products, agrochemicals, and pharmaceuticals. These derivatives serve as important intermediates in the synthesis of various drugs, including indomethacin (a non-steroidal anti-inflammatory drug), dimethylcarbene (an antihypertensive agent), and xylidine (an antihistamine used for treating Alzheimer's disease), among others (Figure 2). Due to its extensive applications, the preparation of indole has been a primary focus of research in organic synthesis since 1866, when Adolf von Baeyer first synthesized indole through reduction with zinc powder.
Numerous traditional methods exist for the synthesis of indole, including the Batcho-Leimgruber method and the Fischer method. However, these conventional synthesis techniques present several drawbacks: they often necessitate the use of costly metal catalysts, which are not only environmentally detrimental but also challenging to eliminate from the final product. Additionally, these methods may require reactions to be conducted under extreme temperature conditions. Consequently, the development of environmentally friendly synthetic routes for indole is the primary focus of our research.
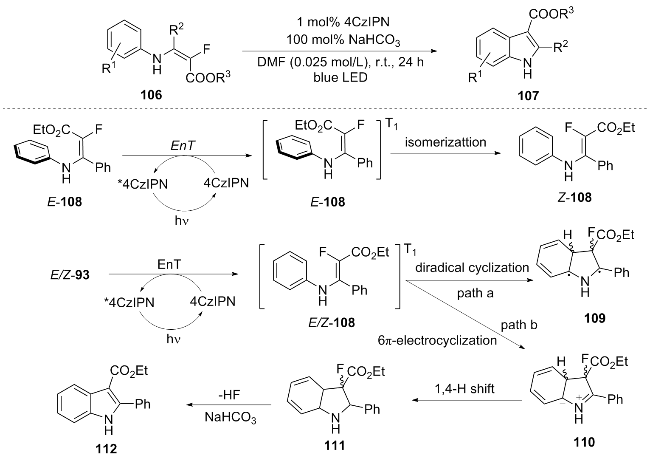
In 2022, Song et al.[24] utilized the energy transfer (EnT) process of the organic photosensitizer 4-CzIPN to achieve the defluorination 6π-cyclization of α-fluoro-β-enamino ester 106 under blue LED light, directly constructing the indole skeleton 107. A reasonable mechanism was proposed. The photosensitizer 4-CzIPN activates the substrate E/Z-93 through triplet energy transfer to generate the triplet [E/Z-108]*; among them, the more planar Z-108* tends to undergo 6π-cyclization (path A), first forming the biradical 109, and then relaxing to the zwitterion 110; while [E/Z-108]* can also directly undergo 6π-electrocyclization to generate 110 (path B), the two paths compete, and ultimately 110 is transformed into the indoline product 112 through 1,4-hydrogen migration (Scheme 14). This methodology offers a metal-free and oxidant-free green synthetic route for the preparation of indoles. The approach demonstrates notable substrate versatility, accommodating electron-donating groups, electron-withdrawing groups, halogens, heterocyclic compounds, and various ester functionalities. Furthermore, it shows potential for gram-scale application with a yield of 90%. However, there remains scope for further improvement in reaction efficiency and the yields associated with certain substrates.
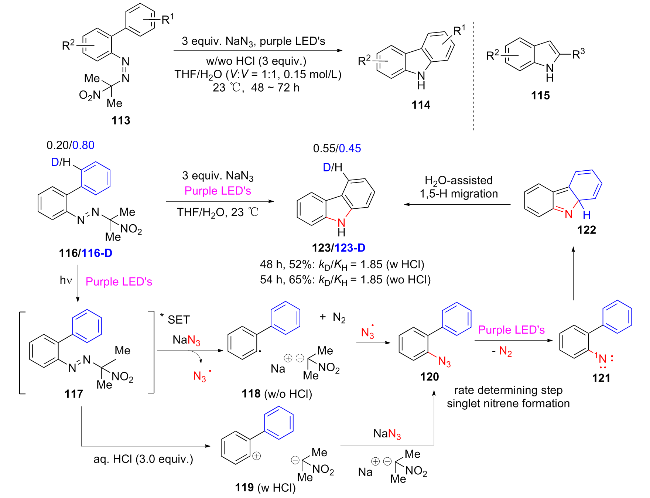
In 2024, Patil et al.[25] developed a one-step synthesis of indole derivatives 115 and carbazole 114 from areneazo- 2-(2-nitro)propanes 113 as the substrate without the need for a photocatalyst, through a visible light-promoted aryl azide formation and intramolecular C—H amination reaction. In the visible light-driven reaction, the substrate 116 first undergoes acid-promoted aryl azide formation (nucleophilic substitution and free radical pathway) to generate azide intermediate 120, which then undergoes photolysis to release nitrile 121 and undergoes intramolecular C—H amination cyclization to form iso-carbazole 122. Finally, through a water-assisted 1,5-hydrogen migration rearrangement, it is efficiently converted into carbazole and indole product 123 in one pot (Scheme 15). This method can be used for the gram-scale synthesis of the natural product Clausine C and can be derivatized into Clausine N/M, showing potential in the fields of medicinal chemistry and green synthesis.
In 2025, Fang et al.[26] utilized the tungstate-free anion (TBADT) as a catalyst to achieve the functionalization of aliphatic C(sp3)—H bonds under near-ultraviolet light irradiation. The aliphatic substrate 109 underwent cyclization with N-aryl acrylamide 110 to efficiently synthesize 3,3- disubstituted oxindole 111. The reaction proceeds via a hydrogen atom transfer (HAT) mechanism to generate alkyl radicals, combined with a K2S2O8 oxidation cycle, to construct the oxindole skeleton 117 under mild conditions (Scheme 16). The anti-inflammatory activity of some products was also verified. The substrate compatibility includes cycloalkanes, toluene, alcohols, and multi-subs- tituted N-aryl acrylamides. The yield of this method at the gram scale is 59%. However, K2S2O8 is required, resulting in low atomic economy. The oxidation system needs to be optimized in the future.
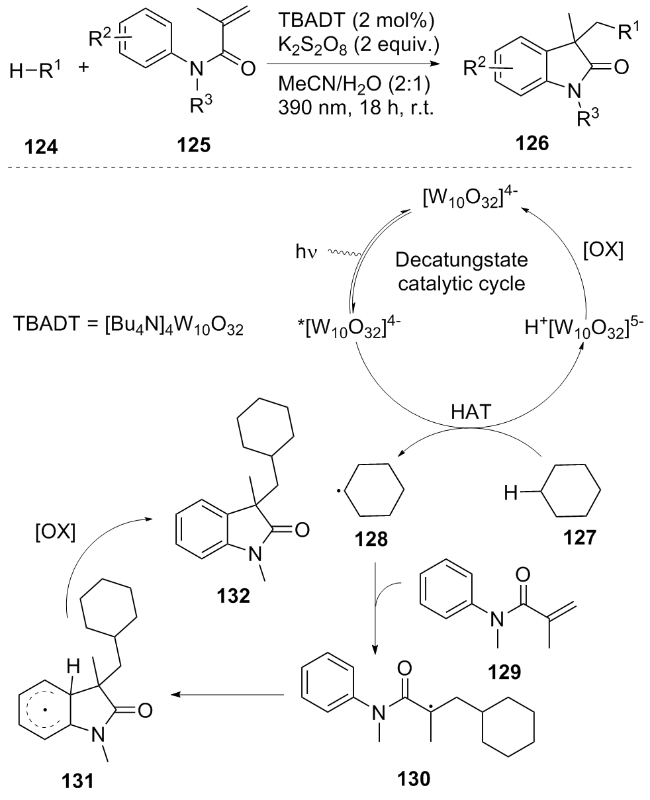
In the same year, Chen et al.[27] developed a visible light-catalyzed three-component radical sulfonylation reaction, which synthesized γ-keto-sulfonyl-substituted oxindole derivatives 120 in one step through a tandem reaction of N-aryl acrylamide 133, cyclopropanol 134 and SO2. A reasonable mechanism was proposed. Under blue light irradiation, the photocatalyst PC was excited to PC*, and the cyclopropanol 134 was oxidized by K2S2O8 or through SET to generate alkoxy radical 136, which underwent β-breakage and ring-opening to form alkyl radical 137, and captured SO2 to form γ-carbonyl sulfonyl radical 138, which added to the alkene 133 and cyclized to form intermediate 140, Finally the product was efficiently constructed through oxidation deprotonation (Scheme 17). Meanwhile, K2S2O8 oxidized PC·⁻ to regenerate the catalyst. This substrate is compatible with a variety of substituted N-aryl acrylamides (including drug molecules, such as ibuprofen and .pngibrozil derivatives) and cyclopropanols. The yield reached 68% in gram-scale synthesis, fully demonstrating the potential of this method in practical applications.
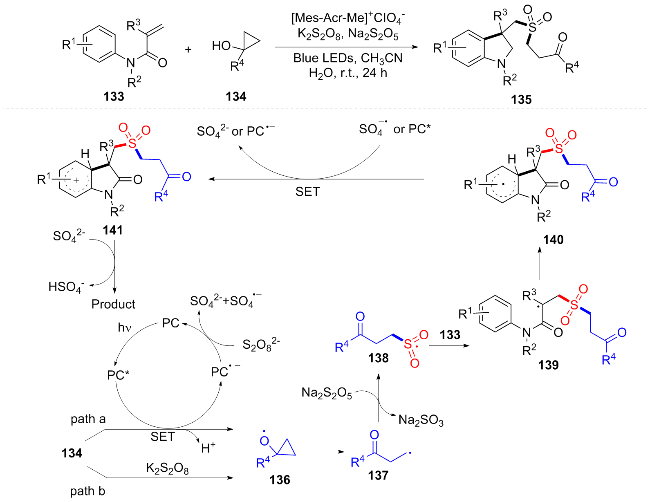
In 2025, Woo et al.[28] also developed a photocatalytic divergent carbon deletion strategy that uses quinoline- N-oxide to photorearrange to generate 3,1-benzooxazine intermediates, which achieve selective carbon deletion in HFIP/toluene solvent systems by reagent-controlled carbon atom scavengers: indoline selectively captures C3 atoms to generate indole directly (mechanism as in Scheme 18B), while aminoethanol excises C2 atoms through intramolecular cyclization to synthesize indole (mechanism as in Scheme 18C). The method (Scheme 18A) provides a new way for the synthesis of indole and nitroindoles, but it is limited by the type of C2 substituent (requiring aryl or strong electron absorbing groups), and the C3 deletion yield of 4-substituted quinoline is low.
Compared with traditional indole synthesis methods that rely on precious metal catalysts and harsh reaction conditions, the photocatalytic strategy enables metal-free syn-thesis under mild conditions, offering improved atom eco- nomy and aligning with the principles of green chemistry.
4-CzIPN (Song, 2022[24]): This system exhibits broad substrate universality, being compatible with electron-do- nating and electron-withdrawing groups, halogens, and heterocyclic compounds. It achieves a 90% yield at the gram scale; however, the efficiency for certain substrates requires further optimization. The reaction is initiated via energy transfer (EnT)-mediated 6π-cyclization.
No photocatalytic system (Patil, 2024[25]): It allows for the gram-scale synthesis of the pharmaceutical compound Clausine C and its derivatives, but displays notable substrate limitations.
TBADT (Fang, 2025[26]): This system enables aliphatic C(sp3)—H functionalization with a 59% yield at the gram scale, although its atom economy is relatively low. The reaction is primarily governed by the hydrogen atom transfer (HAT) mechanism.
[Mes-Acr-Me]+$\text{ClO}_{\text{4}}^{}$. (Chen, 2025[27]): It facilitates a three-component sulfonation reaction compatible with drug-modified acrylamides, including ibuprofen derivatives, achieving a 68% yield at the gram scale and demonstrating promising potential in pharmaceutical synthesis through a radical addition/cyclization pathway.
Divergent carbon deletion (Woo, 2025[28]): Converts quinoline-N-oxides to indoles/nitroindoles via photorearrangement to 3,1-benzooxazine intermediates, using reagent-controlled scavengers (indoline for C3 excision, aminoethanol for C2 excision).
Photocatalytic methods eliminate metal residues, operate under mild conditions, and gram-scale synthesis along with drug-related applications confirms their industrial applicability. Nevertheless, challenges remain, including substrate specificity, reliance on oxidants, and suboptimal efficiency in certain systems.
3 Light-driven catalytic synthesis of benzimidazole and its derivatives
Benzothiazoles and their derivatives represent a significant class of nitrogen-containing heterocyclic compounds, exhibiting a diverse range of applications in pharmacy, industry, and agriculture. These compounds serve as valuable organic molecular backbones, functioning as fundamental structural units in biologically active molecules. In recent years, there has been notable progress in the pharmaceutical applications of benzothiazole and its derivatives.[29-31] For instance, thiramide hydrochloride is employed as a non-steroidal anti-inflammatory drug for treating asthma in both adults and children, while riluzole is primarily utilized as an anticonvulsant for the management of amyotrophic lateral sclerosis (Figure 3). The synthetic development of benzothiazole and its derivatives holds considerable research value.
Classical benzothiazoles and their derivatives have been synthesized through various methods. These methods include the condensation of 2-aminothiophene with substituted nitriles,[32] as well as the cyclization of thiophene phenylaniline[33-34] or 2-halothiophene phenylaniline.[35-36] However, these approaches often present several drawbacks, such as the use of metal catalysts, stoichiometric or excessive amounts of strong oxidizing agents, extended reaction times, and toxic solvents, which are neither economically viable nor environmentally friendly. Therefore, there is a pressing need to develop greener and more efficient methods for the synthesis of benzothiazoles and their derivatives.
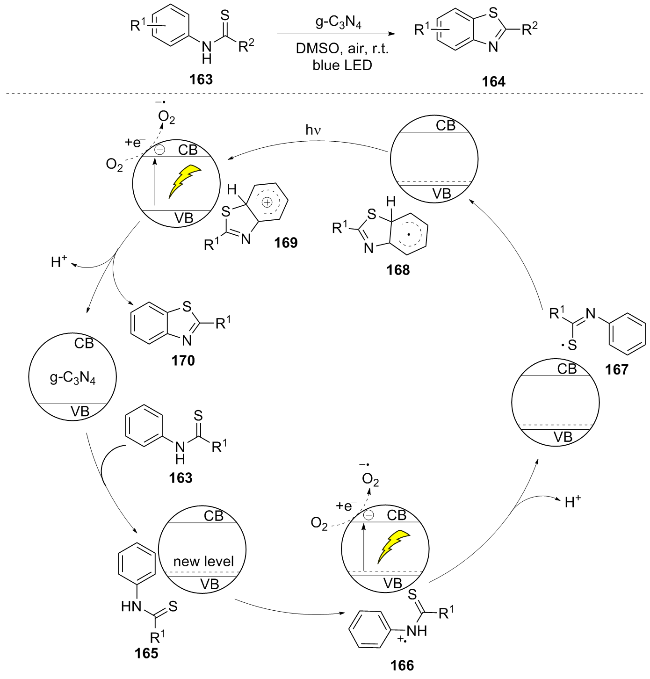
In 2021, Bai et al.[37] developed a heterogeneous photocatalytic system based on graphitic carbon nitride (g-C3N4) for the intramolecular C—H thiolation cyclization of thioacetamide derivatives 163 under ambient air, room temperature, and visible light irradiation, efficiently synthesizing 2-substituted benzothiazole compounds 164. The reaction mechanism involves the adsorption of substrate 163 onto g-C3N4 to form 165, followed by photoexcitation to generate a radical cation 166. This is quenched by O2 to release an electron and deprotonate to form a sulfur radical 167. An intramolecular cyclization then forms an aryl radical 168, which releases an electron to the g-C3N4 conduction band to produce a cation 169. Finally, deprotonation and aromatization yield the benzothiazole product 170 (Scheme 19). This method employs a non-metallic and recyclable g-C₃N₄ catalyst (maintaining a yield of over 93% after five cycles), with the yield in gram-scale experiments dropping to 62%. Its heterogeneous photocatalytic strategy offers a new approach for the formation of C—S bonds.
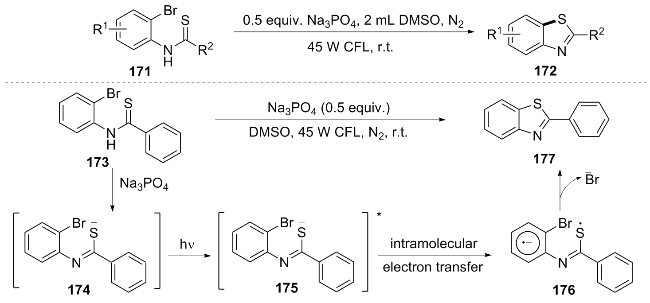
In the same year, Wang et al.[38] achieved a direct dehalo- genation cyclization reaction of o-halothioaniline substrates 171 under mild conditions through visible light irradiation, efficiently generating benzothiazole derivatives 172. The reasonable mechanism of this reaction is as follows: under visible light induction, substrate 174 absorbs photons to generate the excited state 175, followed by intramolecular electron transfer to form intermediate 176, and finally undergoes dehalogenation cyclization to produce product 177 (Scheme 20). This study innovatively utilized the inherent visible light absorption capacity of the substrate to achieve the construction of C—S bonds without the addition of external catalysts. The gram-scale synthesis yield reached 73%, and it was successfully applied to the synthesis of anti-tumor drug precursors (such as B41), further expanding the application prospects of this method in the field of medicinal chemistry.
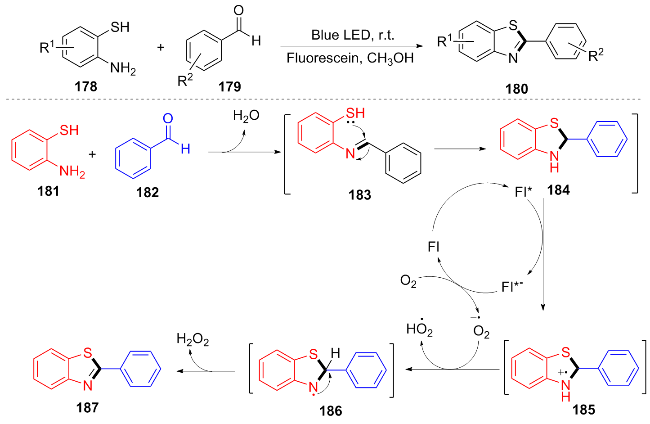
Within that year, Sun et al.[39] used the readily available and inexpensive fluorescein as an organic photocatalyst to carry out the reaction under blue light irradiation and in an air atmosphere. The condensation cyclization reaction between 2-aminophenylthiol 178 and aromatic aldehydes 179 was used to synthesize benzothiazole compounds 180. The plausible reaction mechanism can be described as follows: under blue light irradiation, substrates 181 and 182 undergo nucleophilic addition followed by dehydration to form the imine intermediate 183, which subsequently participates in an intramolecular cyclization to yield compound 184. Within the fluorescein-based system, the excited-state species Fl* donates an electron to 184, generating the radical intermediate 185, while the reduced fluorescein species Fl*⁻ is concurrently oxidized by O2 to regenerate the photocatalyst. Finally, intermediate 185 undergoes deprotonation and hydrogen abstraction to furnish the target product 187 (Scheme 21). This methodology exhibits broad substrate applicability and delivers high yields (82%~94%), effectively addressing the limitations of conventional approaches that rely on transition metals and strong oxidants.
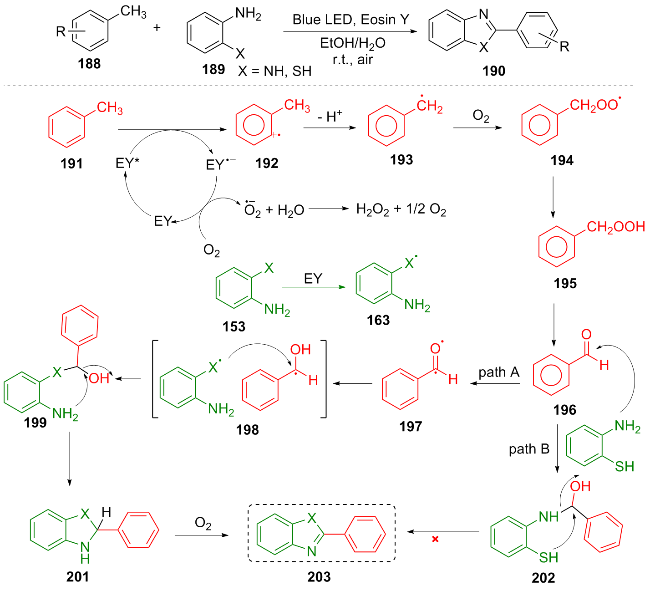
Organic dyes are a very commonly used type of photocatalyst. In 2023, Rajput et al.[40] utilized the organic dye Eosin Y as a photocatalyst. Under blue light irradiation, with methyl aryl compounds 188 and 2-aminothiophenol or 2,4-diaminobenzene 189 as substrates, the C—S and C—N bond formation was achieved through the activation of C—H bonds, efficiently synthesizing benzothiazole and benzimidazole compounds 190. The reaction mechanism is as follows. Blue light excites EY to generate the excited state EY*, which oxidizes 191 through single-electron transfer to form a radical cation 192. The latter loses a proton to form a benzyl radical 193, which is captured by O2 to generate a peroxyl species 194. Through hydrogen abstraction, it is transformed into a hydroperoxide 195, dehydrated to an intermediate 196, and finally condensed with 2-aminothiophenol to yield the target product 203 (Scheme 22). This methodology demonstrates a broad substrate scope, encompassing electron-withdrawing and electron- donating groups as well as heterocyclic rings, and consistently achieves high yields (typically>83%).
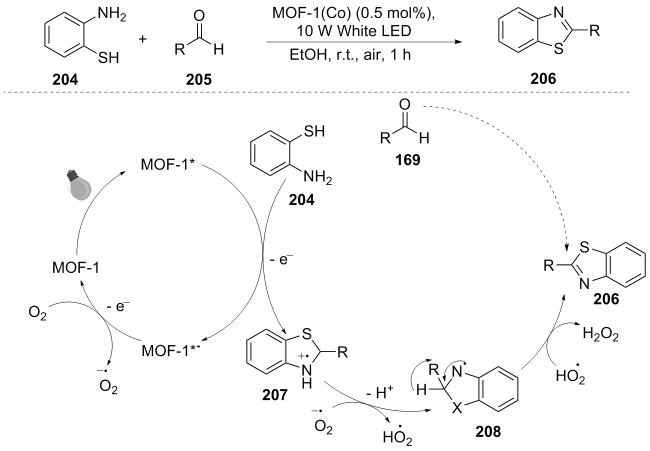
With the rapid development of metal-organic frameworks (MOFs), they have frequently been seen in photocatalysts. In the same year, Liu et al.[41] developed MOFs based on tetrazine chromophores as a recyclable photocatalyst for the synthesis of benzothiazole compounds. The reaction mechanism is as follows. Under light irradiation, MOF-1 (Co) generates the excited state MOF-1(Co)*, while o- aminophenylthiol 168 and benzaldehyde 169 cyclize and reduce to intermediate 170. The excited state catalyst gains electrons to form MOF-1(Co)*⁻, which is oxidized by O2 in the air to regenerate and produce $\text{O}{{_{2}^{}}^{-\ \bullet }}$. Intermediate 172 is ultimately oxidized and dehydrogenated, and undergoes hydrogen abstraction to generate the target product 170 (Scheme 23). This work achieved the efficient and selective synthesis of benzothiazole through the design of a te-trazine-based MOF photocatalyst, highlighting the advan- tages of 2D MOFs in photocatalytic organic transformations and providing a new idea for the development of stable and recyclable heterogeneous photocatalysts.
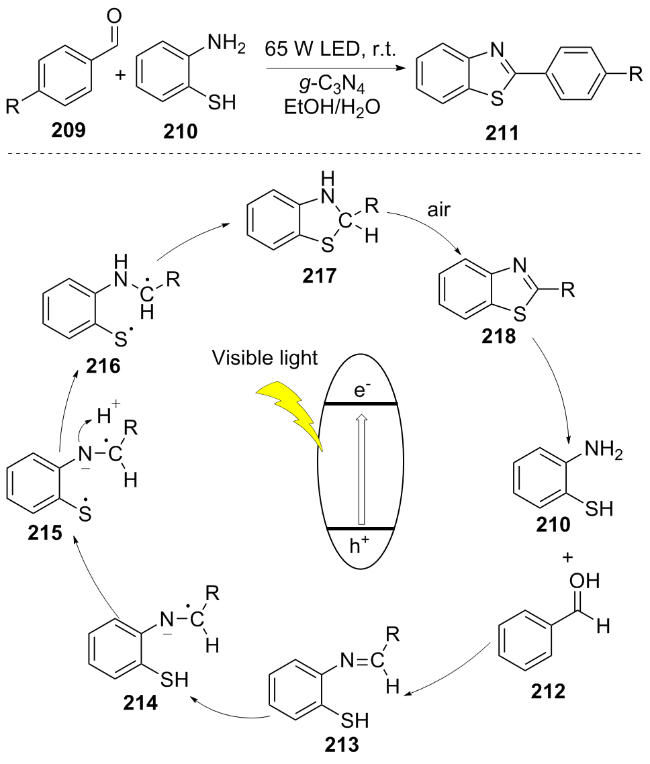
In 2024, Ahl El Haj et al.[42] utilized g-C3N4 as a photocatalyst to catalyze the condensation reaction of aromatic aldehyde 209 with 2-aminophenylthiol 210 under visible light irradiation, synthesizing 2-arylbenzothiazole 211. Under visible light excitation, this reaction generates electron-hole pairs. The photogenerated electrons reduce the imine intermediate 213 to form radical species 214, 215 and 216, which undergo radical coupling to form intermediate 217. Eventually, in the presence of air, dehydrogenation oxidation occurs to yield the benzothiazole product 218 (Scheme 24), while simultaneously achieving the catalytic cycle. This reaction exhibits notable advantages in reaction rate (5-15 minutes) and product yield (89%~97%). More- over, the activity of g-C3N4 remains largely preserved after five reuse cycles, with only a minor decrease observed, thereby further improving the economic viability of the overall process.
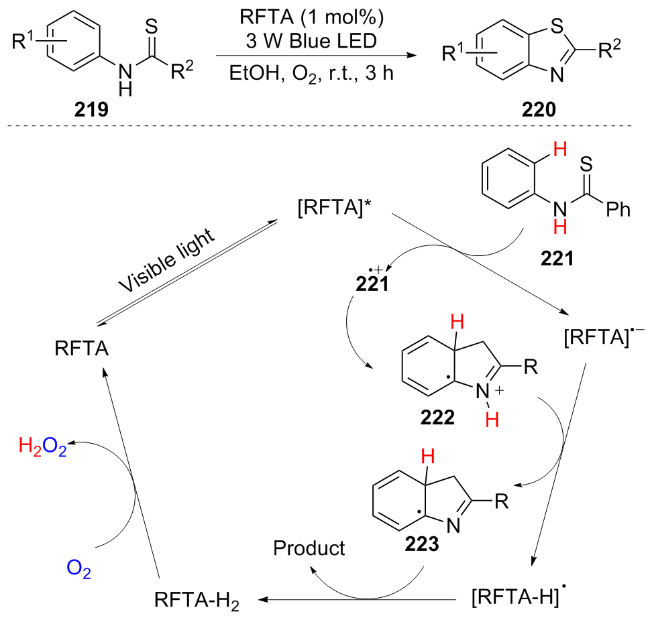
In the same year, Heredia et al.[43] developed a metal-free, base-free, visible-light-driven photocatalytic system based on riboflavin tetraacetate (RFTA) for the efficient synthesis of 2-substituted benzothiazoles 220 in ethanol solvent. The proposed mechanism is as follows: upon light irradiation, RFTA generates the excited state RFTA*, which oxidizes 221 to form the radical cation 221•+, while itself is reduced to RFTA•⁻. The 221•+ undergoes intramolecular cyclization to form 222, and RFTA•⁻ deprotonates 222 to yield the radical 223 and RFTA-H•. These two species undergo a hydrogen atom transfer to generate the product benzothiazole and RFTA-H2. Finally, O2 oxidizes RFTA-H2 to regenerate the catalyst (Scheme 25). This work breaks the traditional reliance on metals and bases. Its mild conditions, excellent functional group compatibility, and simplified operation strategy provide a practical tool for the construction of drug molecules.
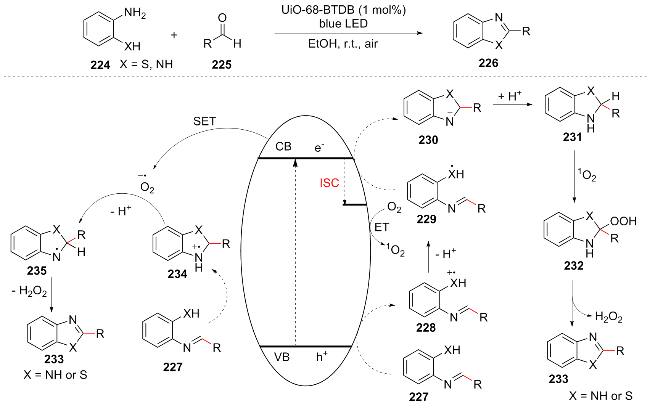
During that very year, Liu et al.[44] designed and synthesized a novel benzothiadiazole-functionalized Zr-MOF (UiO-68-BTDB) as a visible-light-driven efficient ROS generator for catalyzing the condensation cyclization of o-phenylenediamine/o-aminophenylthiol with aromatic aldehydes to synthesize benzimidazole and benzothiazole derivatives. They proposed a reasonable mechanism: under visible light irradiation of UiO-68-BTDB, the two substrates dehydrate to form 227, and the photogenerated holes oxidize 227 to generate 228, which deprotonates to form the radical 229. Meanwhile, the photogenerated electrons undergo intersystem crossing to produce 1O2. After 229 cyclizes, it is reduced by electrons to 230, and then reacts with 1O2 via 231 to release H2O2 and obtain 233. In another pathway, the holes directly cyclize 227 to 234, and the electrons reduce O2 to $\text{O}{{_{2}^{}}^{-\ \bullet }}$. $\text{O}{{_{2}^{}}^{-\ \bullet }}$ promotes the deprotonation of 234 and hydrogen abstraction with 235, ultimately yielding the product 233 (Scheme 26). This work achieved efficient photocatalysis through dual ROS pathways, providing a new strategy for the synthesis of benzoheterocycles.
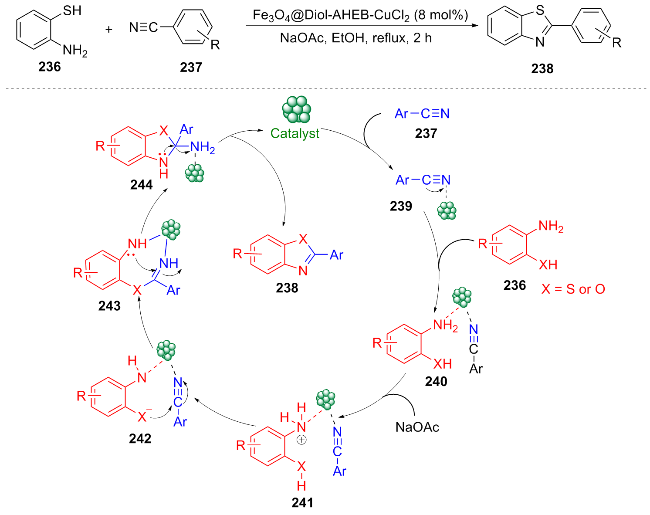
In 2025, Sead et al.[45] developed a novel magnetic recyclable nanocatalyst (Fe3O4@Diol-AHEB-CuCl2) for the synthesis of 2-substituted benzothiazoles 238 through the condensation reaction of aromatic nitriles 237 with 2- aminothiophenol 236 (Scheme 27). This method achieves efficient and selective synthesis of benzothiazoles through a multi-step reaction (imine formation, nucleophilic attack, hydrolysis, and cyclization). The substrate compatibility of this reaction includes electron-withdrawing groups, electron-donating groups, and heterocyclic nitriles. Notably, aliphatic nitriles do not participate in the reaction. The magnetic separation capability and excellent cycling stability (no significant loss of catalytic activity after eight reuse cycles) represent the key advantages of this methodology, offering a novel and sustainable approach for the synthesis of heterocyclic pharmaceutical intermediates.
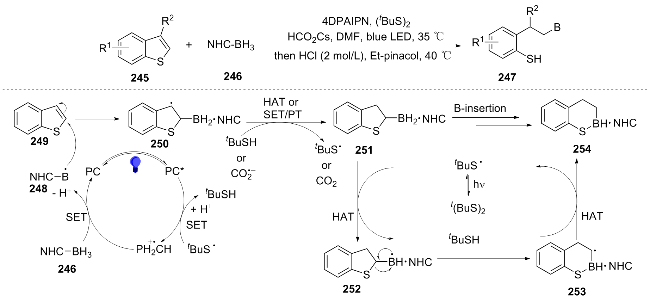
The recently developed photocatalytic boron insertion strategy by Ye et al.[46] enables skeletal editing of benzothiazoles. Under blue light irradiation, this disulfide-media- ted system generates boron radicals from NHC-borane precursors, facilitating simultaneous boron atom insertion into the benzothiazole scaffold and introduction of a formamide group at the nitrogen center. The resulting NHC-stabilized thioborane adduct undergoes hydrolysis to afford ortho-borylated thiophenols—bifunctional molecules bearing tunable thiol and borane moieties. The sulfide unit can be further oxidized, alkylated, or engaged in cyclizations, while the boron center permits reduction or photocatalytic alkylation. Mechanistically, the transformation proceeds via a boron radical-triggered sequence involving β-scission followed by 6-endo-trig cyclization (Scheme 28). This methodology extends to diverse thioheterocycles (e.g., benzothiophenes, thienopyridines) and provides access to pharmacologically relevant building blocks, exemplified by the synthesis of a precursor to the antipsychotic agent brexpiprazole.
Compared with traditional methods that rely on metal catalysts, strong oxidants, and toxic solvents, photocatalytic synthesis of benzothiazole compounds offers notable advantages, including mild reaction conditions, the use of air as a green oxidant, and enhanced environmental compatibility. Various photocatalytic systems exhibit distinct chara- cteristics. g-C3N4 demonstrates broad substrate applicability, being compatible with electron-withdrawing groups, electron-donating groups, naphthalene rings, and alkyl substituents, and it enables gram-scale synthesis. The catalyst-free system has successfully synthesized the anti- tumor drug precursor B41 with a gram-scale yield of 73%, showing good compatibility with halogenated anilines. However, its performance with fluorinated substrates remains limited. Organic dyes (e.g., fluorescein and Eosin Y) offer cost-effectiveness and favorable substrate compatibility, including tolerance for heterocycles and various functional groups, although their recyclability has not been systematically validated. UiO-68-BTDB exhibits high stability and reusability, enabling highly selective synthesis through a dual reactive oxygen species (ROS) pathway, but its MOF synthesis is relatively complex and time-consu- ming. RFTA shows reduced efficiency with substrates containing strong electron-withdrawing groups. Fe3O4@- Diol-AHEB-CuCl2-Fadhel provides convenient magnetic separation and excellent cycling stability, yet the system incorporates copper, a metal component. Ye’s[46] boron insertion enables benzothiazole skeletal editing via blue light/disulfide-generated NHC-borane radicals. It simultaneously inserts boron atoms and installs formamide groups, yielding ortho-borylated thiophenols—bifunctional intermediates amenable to diverse modifications. The reaction proceeds through boron radical-triggered β-scission/6-en- do-trig cyclization. It is applicable to benzothiophenes/thie- nopyridines and synthesizes brexpiprazole precursors.
In summary, photocatalytic methods have enabled the synthesis of benzothiazole derivatives through innovative mechanisms, such as intramolecular electron transfer, radical pathways, and dual ROS synergy. Their successful implementation in gram-scale amplification experiments and direct application in the synthesis of anti-tumor drug precursors further confirm their potential for industrial and practical applications. Future research should focus on enhancing scalability, expanding substrate scope—inclu- ding aliphatic aldehyde substrates—and simplifying catalyst synthesis procedures to promote broader applicability and practical utility.
4 Light-driven catalytic synthesis of triazole and its derivatives
Triazoles consist of three nitrogen atoms and two carbon atoms, and they can be classified into two isomers: 1,2,3- triazole and 1,2,4-triazole. Compounds containing triazole represent a significant class of nitrogen-containing heterocyclic compounds, exhibiting remarkable biological properties, including anticancer,[47-48] antituberculosis,[49-51] antimicrobial,[52] and anti-AIDS[53] activities. Triazole scaffolds serve as crucial pharmaceutical intermediates and are incorporated in various drug formulations, such as antimicrobials like fluconazole and tazobactam, as well as antivirals like ribavirin (Figure 4). Given the vital applications of triazole scaffolds in pharmaceutical intermediates, identifying effective synthetic methods is of paramount importance.
Although triazoles can be skillfully synthesized under conventional reaction conditions, the primary drawbacks of these methods include product stability and reduced yield. Consequently, there is a growing interest in green synthesis utilizing sustainable catalytic conditions.
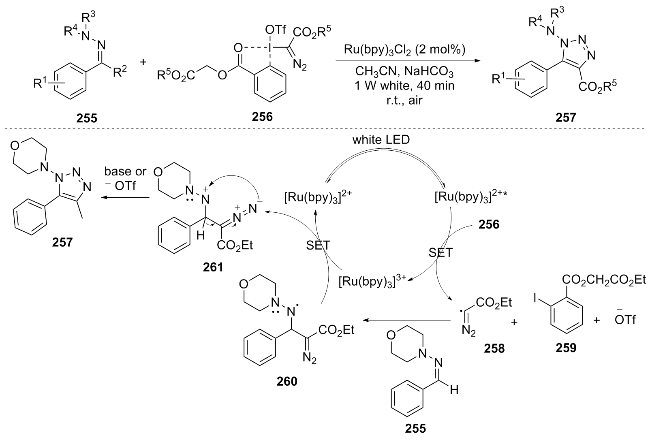
In 2021, Dong et al.[54] developed a visible light-induced [3+2] cycloaddition reaction for the efficient synthesis of 1-amino-1,2,3-triazole compounds 211. The proposed reac-tion mechanism is as follows: under light irradiation, [Ru(bpy)3]2+ is excited to [Ru(bpy)3]2+*, which oxidizes the substrate to generate diazoalkyl radical 212 and [Ru- (bpy)3]3+. Subsequently, 212 attacks the C=N bond to form amino radical 214, which is oxidized by [Ru(bpy)3]3+ to amino cation 215 and regenerates [Ru(bpy)3]2+, completing the catalytic cycle. Finally, 215 undergoes intramolecular cyclization to produce the product 1-amino-1,2,3-triazole 211 (Scheme 29). This method is carried out under mild conditions and has a wide range of substrate applicability. It has been successfully applied to the late-stage functionalization of natural products (such as estrone and estriol) and the synthesis on a gram scale.
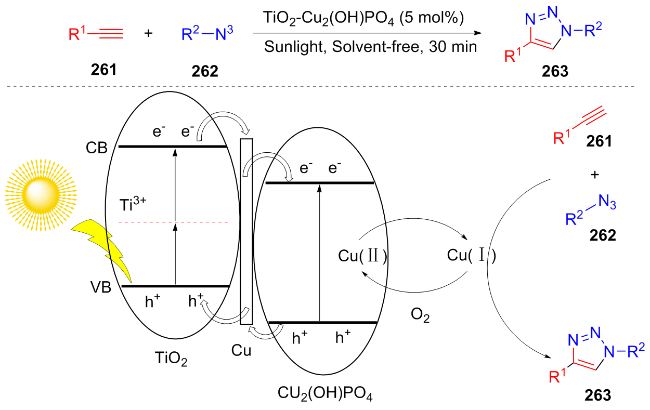
In the same year, Kiranmye et al.[55] utilized TiO2-Cu2(OH)PO4 nanophotocatalyst under solvent-free and sunlight conditions to efficiently synthesize 1,4-disub- stituted 1,2,3-triazole compounds 263 from phenylacetylene 261 and benzyl azide 262. The author proposed a reasonable reaction mechanism: under light irradiation, the electron transition excitation of TiO2 occurs, and the electrons are transferred in situ to Cu(II) via copper to form Cu(I), which drives the azide-alkyne cycloaddition reaction. Subsequently, Cu(I) is oxidized by O2 to regenerate Cu(II), completing the catalytic cycle (Scheme 30). This catalyst can be reused more than six times with little loss of catalytic efficiency, providing a green and sustainable condition for applications in drug and synthetic chemistry.
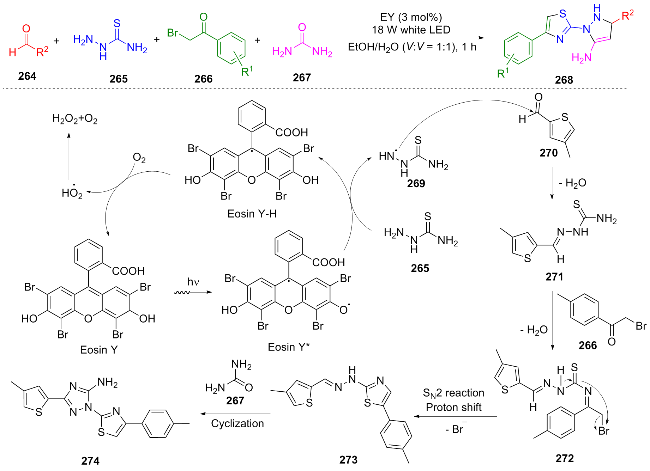
In 2022, Tabassum et al.[56] utilized white light LED irradiation and Eosin Y as a photocatalyst to synthesize 12 novel 1,2,4-triazole-3-amine derivatives 268 through a one-pot sequential addition of substrates (substituted benzoyl bromide 266, aromatic aldehyde 264, aminothiourea 265, and urea 267). Under white light, Eosin Y* catalyzed the transformation of aminothiourea 265 into intermediate 269, which then condensed with aldehyde 270 to form imine 271. Intermediate 271 further condensed with bromide 266 to yield intermediate 272. Through an SN2 elimination of bromine, a thiazole ring was constructed, and finally, urea 267 was cyclized to form the triazole ring, resulting in product 274 (Scheme 31). This method does not require metal catalysts, is simple to operate, and achieves high yields (83%~94%), conforming to the principles of green chemistry.
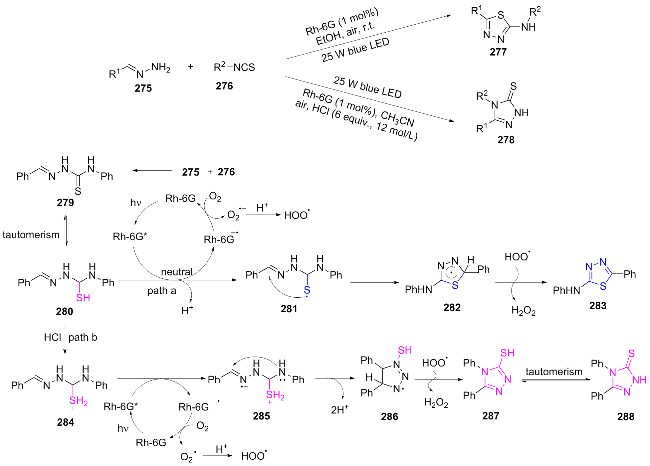
In 2023, Teng et al.[57] developed an acid-regulated photocatalytic divergent reaction, which enabled the selective synthesis of two types of heterocyclic compounds from the same substrates (275 and 276): 2-amino-1,3,4-thiadia- zole 277 under neutral conditions and 1,2,4-triazole-3- thione 278 under acidic conditions. The mechanism of this reaction is as follows: 275 reacts with 276 to form 279, which undergoes tautomerization to 280. Under irradiation, Rh-6G is excited to Rh-6G*. Path A: 280 reacts with Rh-6G to generate a sulfur radical 281, which cyclizes to 282 and then undergoes hydrogen transfer to form 283. Path B: In acid, 280 converts to 284, and Rh-6G is oxidized to a nitrogen radical 285 (Scheme 32). Cyclization and hydrogen transfer yield 288, while Rh-6G is oxidized by O2 and regenerated, completing the catalytic cycle. This reaction can be conducted at room temperature in an ambient air atmosphere under blue LED irradiation, without requiring metal catalysts or chemical oxidants. It exhibits high selectivity and broad substrate applicability. A gram-scale synthesis was successfully demonstrated, achieving a yield of 80%.
Traditional methods frequently face challenges such as poor product stability and low reaction yields. In contrast, photocatalytic synthesis enables green chemical transformations under mild reaction conditions, significantly improving both reaction efficiency and atom economy. The photocatalytic synthesis of triazole compounds demonstrates relatively broad substrate universality, accommodating various functional groups including halogens (F, Cl, Br), cyano, trifluoromethyl, alkoxy groups, as well as aromatic substrates such as substituted benzaldehydes and aryl azides. Certain methodologies have successfully achieved gram-scale synthesis and the modification of natural products. A common mechanistic core among these approaches involves the excitation of the photocatalyst, which initiates electron transfer and generates radical or cationic intermediates to drive cyclization. The catalyst is subsequently regenerated with the assistance of molecular oxygen. However, existing systems still face limitations, including reliance on precious metals (e.g., Ru), restricted substrate scope (e.g., TiO2-Cu system), and complex reaction condition control (e.g., Rh-6G system).
5 Light-driven catalytic synthesis of pyrrolidines and its derivatives
Pyrrolidine is a unique nitrogen-containing backbone found in a variety of biologically active natural products and top-selling pharmaceuticals. It is the most prevalent five- membered non-aromatic nitrogen heterocycle among FDA- approved drugs, including asenapine (an antipsychotic) and ethosuximide (an antiepileptic) (Figure 5). Pyrrolidine is highly favored in the fields of pharmaceutical sciences and drug design. Furthermore, pyrrolidine and its derivatives are extensively utilized as transition metal ligands and organocatalysts in asymmetric synthesis, where they play a crucial role in controlling stereoconformations with high selectivity. The promising applications of pyrrolidine have motivated chemists to investigate new synthetic methods.
In 2021, Li et al.[58] developed a metal-free, visible-light-driven triiodide-mediated [3+2] cycloaddition reaction for the efficient synthesis of polysubstituted pyrrolidines 291. This reaction utilized the inexpensive and stable tetra-n-butylammonium iodide (TBAI) as a radical initiator, and in reagent-grade NMP solvent, the in-situ generated triiodide (TBAI3) was excited by visible light to achieve the cycloaddition of N-tosyl aziridines 290 with alkenes 289. The reaction exhibited excellent regioselectivity and good functional group compatibility. They proposed a possible reaction mechanism: under light irradiation, TBAI3 promoted the ring-opening of 290 to form alkyl radical 292, which added to phenethyl 289 to form 293, underwent intramolecular rearrangement to form amino radical 294, and then extracted hydrogen from the solvent to form iodide 295, and finally underwent intramolecular cyclization to form the pyrrolidine product 291 (Scheme 33). This method is applicable to aryl, heteroaryl alkenes, polyfluoroarenes and bioactive molecules (such as estrone, glucose derivatives), showing good application prospects, but the reaction needs to be carried out at 70 ℃ for 8 h.
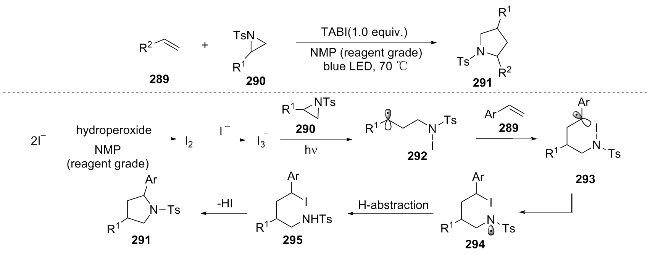
In the same year, Wei and his colleagues[59] achieved a one-step cyclization of styrene 296, α-bromoalkyl esters 297 and primary amines 298 in a microchannel reactor using visible light-activated photocatalysts to synthesize pyrrolidin-2-one 299. The proposed reaction mechanism is as follows: visible light excitation of PC-B* initiates the cleavage of the brominated compound to form free radical 301, which adds to styrene 302 to yield 303. This intermediate is oxidized by PC-B+ to a carbocation and reacts with the primary amine to give the product 307. PC-B+ is then reduced to regenerate the catalyst, completing the cycle (Scheme 34). Compared with traditional reactions, microfluidic technology significantly enhances the reaction efficiency and suppresses side reactions.
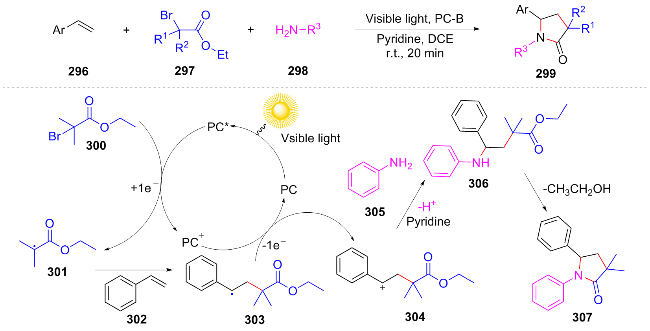
In 2023, Mao[60] efficiently synthesized fluorinated pyrrolidine derivatives through a cascade transformation of fluoroalkyl iodides and N,N-diallylamine. The plausible mechanism is as follows: compound 308 forms a halogen bond complex with fluoroalkyl iodides, generating fluoroalkyl radicals under blue LED irradiation. Subsequently, this radical selectively undergoes tandem addition with 308 to form a carbon radical intermediate 312, which abstracts an iodine atom from RFI to yield the target product and regenerate the RF radical (Scheme 35). This reaction successfully modified amino acids and drug molecules, providing a new approach for the development of fluorinated drugs.
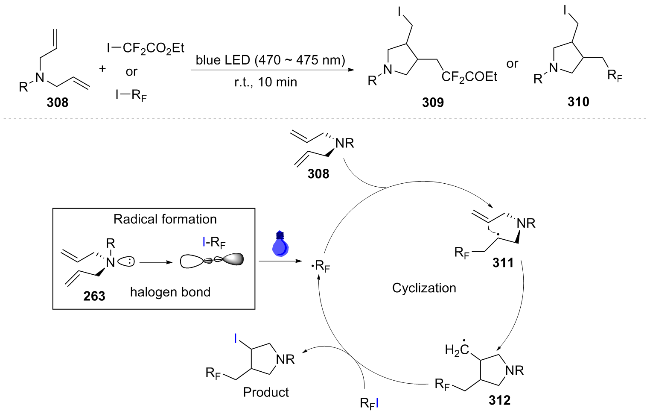
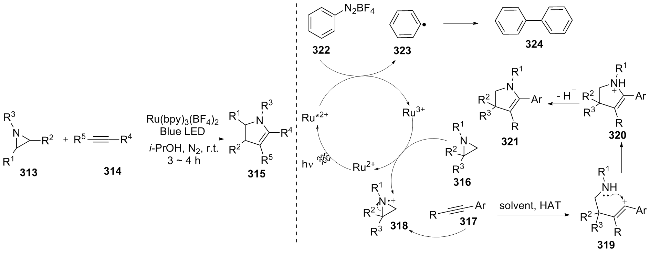
In the same year, Siddiqui[61] also conducted a visible light-catalyzed [3+2] cycloaddition reaction, using aziridine 313 and activated alkynes 314 as substrates, and efficiently synthesized multi-substituted dihydropyrrolidines 315 under the catalysis of Ru(bpy)3(BF4)2 and blue light irradiation. Visible light excitation of the Ru catalyst oxidized 322 to generate phenyl radicals 323, which dimerized to 324. Meanwhile, Ru3+ oxidized aziridine 316 to radical cation 318 (Ru3+ was reduced and regenerated), and 318 was attacked by alkane 317 to form intermediate 319, which underwent intramolecular cyclization and HAT to yield product 321 (Scheme 36). This work provided a new approach for the synthesis of dihydropyrrolidines, but the reaction's progress relied on an oxidant, and the influence of strong electron-withdrawing and electron-donating groups on the substrates had not been studied.
Within that year, Zhou[62] developed a mild and efficient aerobic decarboxylation [2+2+1] cycloaddition reaction under organic photocatalysis. This reaction utilized 3-substituted chromones 325 and N-aryl glycines 326 as substrates to synthesize pyrrolidines 327 under blue light irradiation. They proposed a reasonable mechanism: visible light excitation of 4CzIPN was reduced and quenched by 328 to generate the radical cation 329, which deprotonated and decarboxylated to form the aminomethyl radical 330, which underwent Giese addition with 326 to form 331 and was reduced and protonated by 4CzIPN•− to form 332. Meanwhile, the reactive oxygen species generated by the oxidation of 4CzIPN•− by O2 oxidized 330 to the imine 333, and 332 nucleophilically attacked 333 to form the iminium 335, which underwent an intramolecular Mannich reaction to yield the product (Scheme 37). The photocatalytic [2+2+1] cycloaddition reaction for the construction of the pyrrolidine ring avoids the dependence on formaldehyde or activated ketones in traditional [3+2] cycloaddition reactions. Its mild conditions provide a new tool for medicinal chemistry.
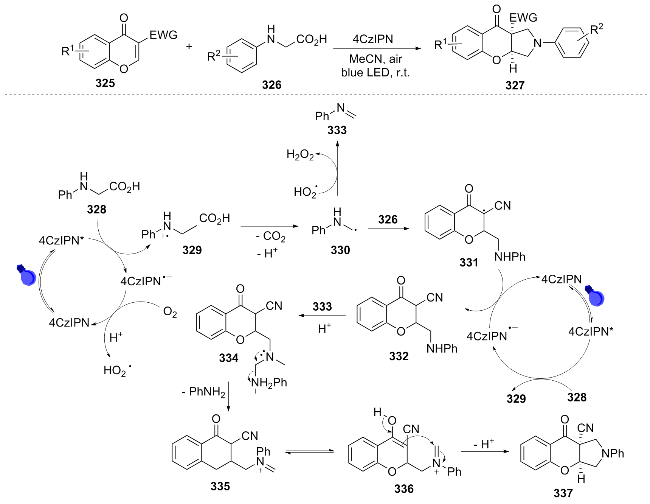
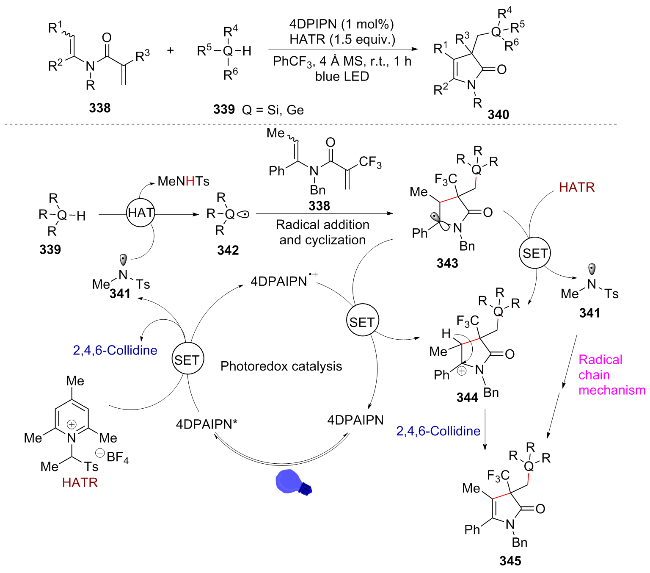
In 2024, Bajya[63] efficiently synthesized trifluoromethylated 4-pyrroline-2-ketone derivatives 340 containing silicon- or germanium-based functional groups through a regioselective radical addition followed by a cascade cyclization reaction between N-alkenyl α-trifluoromethyl-acrylamides and silanes or germanes. They proposed that the reaction mechanism involves visible light excitation of 4DPAIPN* to initiate a SET process with HATR, resulting in the formation of the sulfonamide radical 341 (accompanied by the release of a base). This radical subsequently undergoes hydrogen atom transfer (HAT) with a silane or germane to generate radical 342, which proceeds through addition and cyclization to afford compound 343. The main pathway: 344 is oxidized by HATR to regenerate 341 (chain cycle) to produce the product 345. The secondary pathway: 343 undergoes SET oxidation with 4DPAIPN• to form 344, which is deprotonated to yield the product 345 (Scheme 38). This reaction demonstrates high synthetic value due to its mild reaction conditions and good compatibility with various functional groups. Notably, this method can be applied to the synthesis of drug molecules (such as ibuprofen and naproxen), and it also supports the functional group modification of molecules in the later stage.
Compared with traditional pyrrolidine synthesis methods that rely on harsh conditions such as high temperatures, metal catalysts, or strong oxidants, the photocatalytic strategy offers significant advantages, including mild reaction conditions, enhanced environmental compatibility, and reduced reliance on metallic reagents. In recent years, photocatalytic approaches to pyrrolidine synthesis have demonstrated excellent performance in the modification and functionalization of aromatic substrates. However, their applicability to aliphatic substrates remains limited. Notably, certain methodologies exhibit promising industrial potential due to their efficient scalability and successful application in drug molecule synthesis. The reaction mechanism predominantly follows radical pathways, with the oxidation strategy playing a crucial role in determining overall reaction efficiency.
6 Light-driven catalytic synthesis of pyrimidines and its derivatives
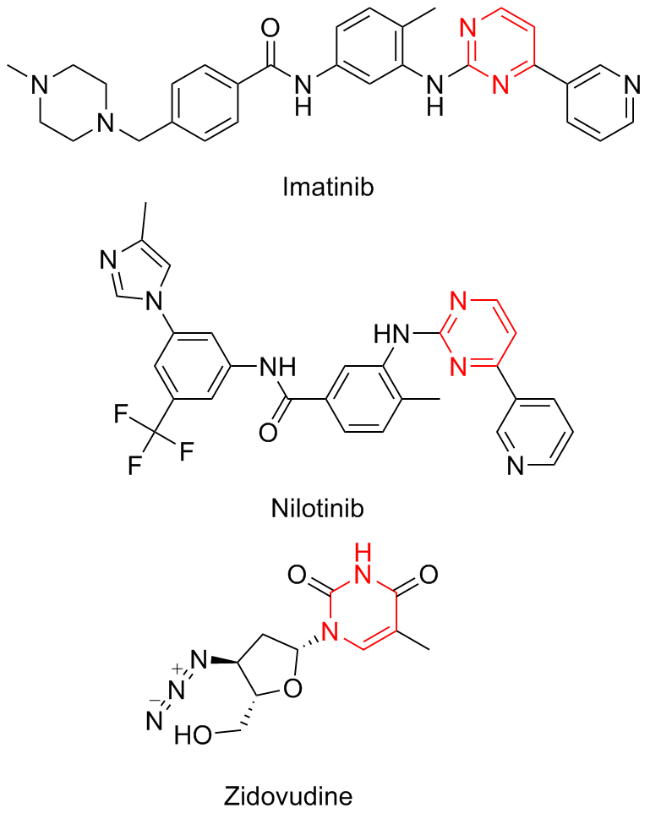
Pyrimidines are heterocyclic compounds characterized by the presence of two nitrogen atoms. They serve as a fundamental structural backbone in various natural products, agrochemicals, and pharmaceuticals.[64,65] Notable examples include imatinib and nilotinib, which are used in the treatment of chronic granulocytic leukaemia, as well as zidovudine, an antiviral agent (Figure 6). Furthermore, numerous pyrimidine derivatives exhibit a range of biological activities, including antibacterial,[66] anti-inflammatory,[67] anticancer,[68-69] as well as antipyretic and analgesic[70] effects. Given the extensive applications of pyrimidine derivatives in medical and biological fields, their synthesis has garnered significant attention.
Classical approaches to the synthesis of pyrimidine derivatives include transition metal-catalyzed condensation, cyclization, or oxidation of amides with 1,3-dicarbonyl derivatives, saturated ketones, α,β-unsaturated ketones, ethyl ketones, and allyl-aryl compounds. However, these methods typically require multiple reaction steps and the use of costly metals. Despite the emergence of numerous methods for constructing multisubstituted pyrimidine derivatives over the past few decades,[71-72] the development of new, efficient, and environmentally friendly methods for the synthesis of pyrimidine derivatives remains a significant area of research. This includes light-driven approaches to the construction of pyrimidines and their derivatives.
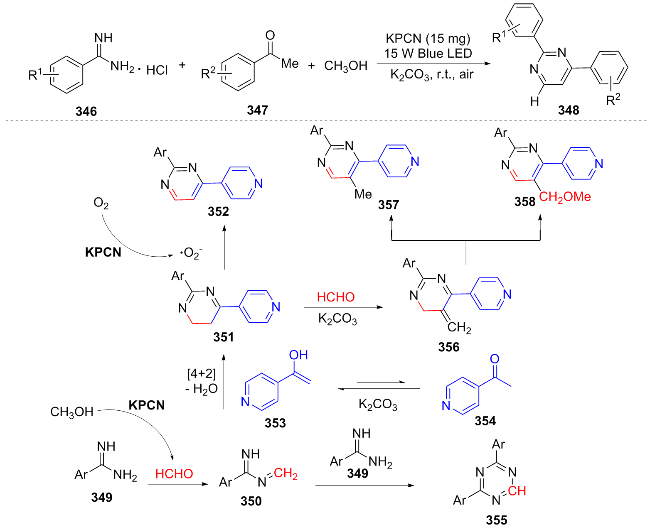
In 2024, Wu[73] utilized potassium ion-modified polymeric carbon nitride (KPCN) as a catalyst to achieve the synthesis of pyrimidines and their deuterated derivatives through the coupling of methanol oxidation and oxygen reduction reactions. They proposed a mechanism: under visible light, KPCN generates electron-hole pairs; electrons reduce O2 to form superoxide radicals, and holes oxidize alkanols to aldehydes. These aldehydes condense with 349 to form iminium 350. Under the action of a base, the enol form of ketone 353 undergoes [4+2] cycloaddition with 350 and dehydration to form intermediate 351, which is oxidized to the main product 352 (Scheme 39). Some 351 reacts with HCHO under K2CO3 to form 356, which tautomerizes to 357 or reacts with methanol to yield the by-product 358. This work provides a green new route for the synthesis of pyrimidines, especially demonstrating significant potential in the synthesis of deuterated drugs. It has broad substrate compatibility (both aryl, heteroaryl, and alkyl substrates are compatible), and the yield of gram-scale synthesis is 66%, showing potential for industrial application.
In 2025, Zhu[74] developed a one-pot photocatalytic synthesis strategy to convert aldehyde 359 into 5-substituted 4-perfluoroalkylpyrimidine derivatives 363. The reasonable mechanism of this reaction is as follows: the in-situ generated enal 365 undergoes 1,2-addition to form 366, and its four isomers are converted to Z,Z-366 under guanidine catalysis and then undergo intramolecular cyclization to yield the target product 367. In the parallel path, 365 undergoes 1,4-addition to form 368, which eliminates HF to become 369 and can condense with 367, or 368 mutarotates to 370 and condenses to form heterocycle, which then eliminates HF and undergoes aromatization to give the final product (Scheme 40). This method is compatible with a variety of functional groups (such as aliphatic aldehydes, aromatic aldehydes, and aldehydes containing ester, hydroxyl, or amino protecting groups), and has been successfully applied to the derivatization of bioactive molecules (such as amiloride). However, this reaction requires a relatively long reaction time and a relatively high reaction temperature.
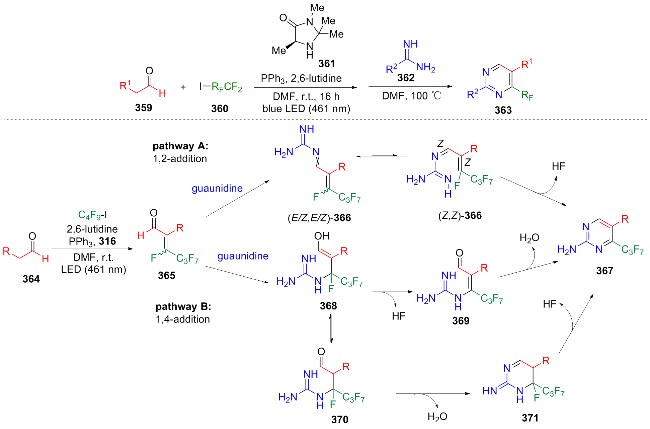
Over the past five years, relatively few studies have reported on the photocatalytic synthesis of pyrimidines; however, these approaches have demonstrated significant innovation. In comparison to traditional transition metal-catalyzed condensation or cyclization methods—which require multiple synthetic steps, costly metals, and harsh reaction conditions—photocatalytic strategies have achieved notable breakthroughs through the use of mild reaction conditions, streamlined synthetic procedures, and environmentally sustainable processes.
7 Light-driven catalytic synthesis of quinolines and its derivatives
Quinolines, also known as benzopyridines, are nitrogen-containing heterocyclic organic aromatic compounds. Quinoline is prevalent in numerous natural products and serves as an important structural scaffold in various drugs. For instance, it is a key component in bedaquiline, an anti-tuberculosis medication, and camptothecin, an anti-tumor agent (Figure 7). Furthermore, a diverse array of biological. activities associated with quinoline and its derivatives has been documented, including antibacterial,[75-76] antiviral, [77] and anti-Alzheimer's disease [78] properties. Consequently, the development of efficient and environmentally friendly synthetic methods for quinoline and its derivatives has become a primary focus of current research.
One of the most reliable conventional methods for the preparation of quinoline is the Povarov reaction-oxidation. The Povarov reaction is catalysed by protonic or Lewis acids and involves the cycloaddition of azodienes and dienophilic reagents in the form of [4+2] to produce tetrahydroquinoline. Subsequently, the formed tetrahydroquinoline is oxidised to quinoline. Where the oxidation step usually requires the use of stoichiometric amounts of oxidants such as peroxides, high valence metals, tetramethylpiperonyloxy (TEMPO) oxyammonium salts and iodine (III) oxidants.[79-82] This results in low economy of the reaction and is less desirable from the point of view of green chemistry. So development of green and economical synthetic methods is necessary.
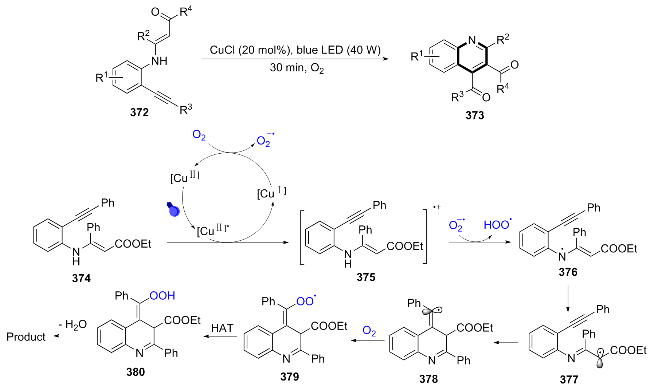
In 2021, Huang[83] developed a mild and efficient visible light-induced, copper-catalyzed oxidative cyclization reaction, which achieved the intramolecular cyclization of substituted aromatic enaminones with the C(sp3)—H bond adjacent to nitrogen, successfully synthesizing 373, a multi-substituted quinoline. Visible light excitation of Cu(II) led to a single-electron transfer with substrate 374, generating Cu(I) and radical 375. Cu(I) was oxidized by O2 to regenerate Cu(II), simultaneously producing $\text{O}{{_{2}^{}}^{-\ \bullet }}$. Radical 375 underwent hydrogen transfer with $\text{O}{{_{2}^{}}^{-\ \bullet }}$ to form HOO• and resonance structures 376 and 377. Radical 377 underwent intramolecular radical cyclization to yield 378, which was captured by O2 and underwent hydrogen transfer to form peroxide 380, and dehydration gave the product (Scheme 41). This method is applicable to o-amino-phenylacetylene derivatives containing electron-donating groups, electron-withdrawing groups, heterocycles, and alkyl groups.
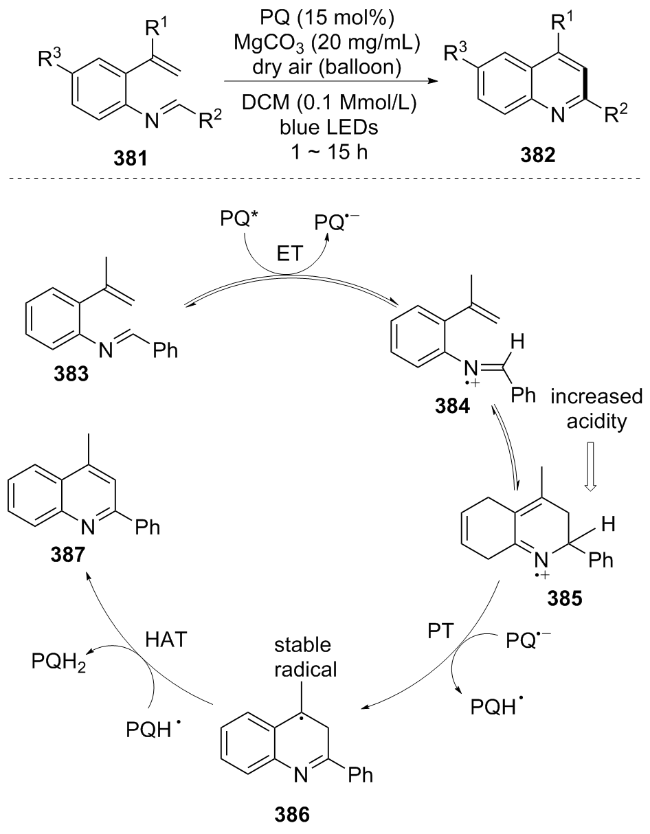
In 2022, Talvitie[84] used 2-vinyl aniline 336 as the substrate and achieved electrocyclization through a photo-catalytic process sensitized by phthalic anhydride(PQ), efficiently synthesizing multi-substituted quinoline 382. The reaction mechanism can be briefly described as follows: visible light excites the photosensitizer PQ to PQ*, initiating a single-electron transfer from 383 to generate the nitrogen radical intermediate 384, which undergoes cyclization to form the cationic radical 385. PQ•− abstracts a hydrogen atom from 385 to generate the neutral radical PQH• and 386. Finally, a hydrogen atom transfer occurs to produce the product quinoline 387 and the phenolic PQH2, which is oxidized by oxygen to regenerate PQ (Scheme 42). This method is compatible with a variety of aryl-substituted substrates but has limitations with aliphatic-substituted substrates. Additionally, PQ is a cheap organic photocatalyst and is more accessible than noble metal catalysts.
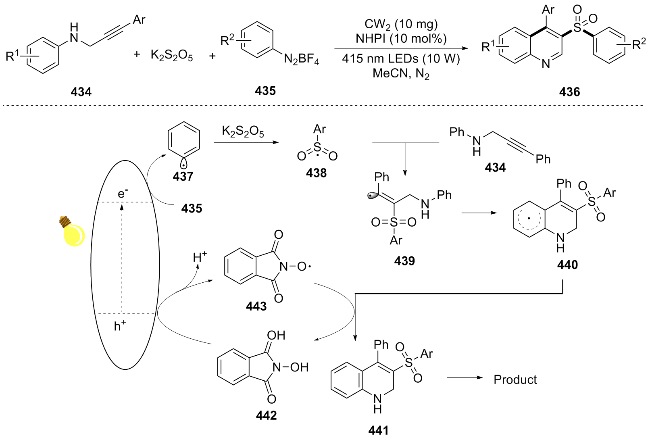
In the same year, Su[85] developed a dual-catalyst system (photocatalyst and proton reduction cocatalyst) to achieve the efficient synthesis of quinoline 389 using N-alkylaniline 388 or aniline 390 and aldehyde 391 as substrates. This reaction proceeds through the synergistic action of the photocatalyst and cobalt catalyst: the excited-state Ru2+ transfers electrons to Co3+ to generate Ru3+ and Co2+, and Ru3+ oxidizes N-alkylaniline to produce a radical cation, which deprotonates to form an α-amino radical; this radical is oxidized by Co2+ to an imine ion, which hydrolyzes to an imine and isomerizes to an enamine; the two undergo a [4+2] cycloaddition via the Paal-Knorr reaction to form a tetrahydroquinoline skeleton, and then deprotonate aniline to form dihydroquinoline, and finally undergo photoreductive dehydrogenation aromatization to generate the quinoline product (Scheme 43). This method demonstrates broad substrate generality and circumvents the reliance on stoichiometric oxidants in traditional methods. However, the cost of the precious-metal catalyst remains high.
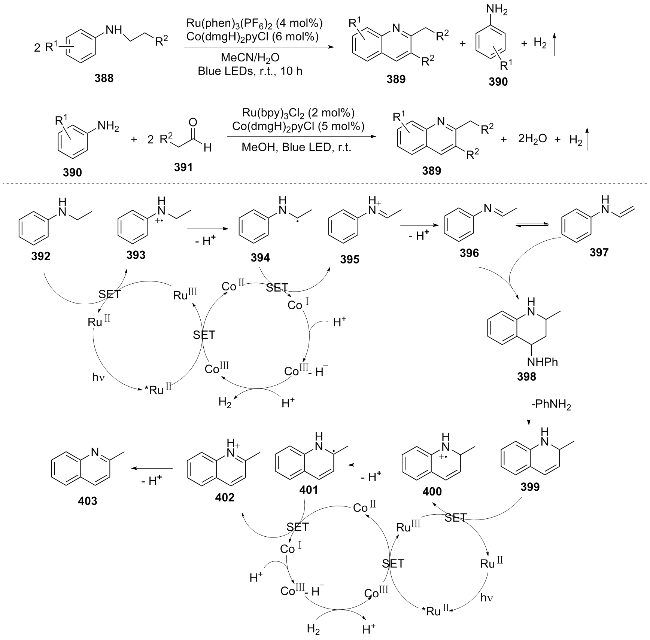
Due to the economic nature of organic dyes, they have been chosen by many researchers as photocatalysts. In 2023, Jaiswal[86] used Eosin Y as a photocatalyst to efficiently synthesize quinoline derivatives 406 through a tandem reaction of styrene oxide 404 and aniline 405 at room temperature under visible light irradiation. Visible light excitation of Eosin Y undergoes a double quenching cycle to generate the active intermediate 412, which dehydrates and hydrolyzes to form intermediate 414. Under light, this intermediate undergoes homolytic cleavage to produce radicals 415. Radical 415 undergoes a 1,3-shift and combines with 417, eliminating benzaldehyde, and then undergoes intramolecular cyclization to form intermediate 420. Finally, in the presence of oxygen, 420 undergoes imine-enamine tautomerization and aromatization to yield the product (Scheme 44). This methodology offers several advantages, including mild reaction conditions, high product yields (up to 96%), short reaction times (30 minutes), excellent atom economy, and broad substrate applicability.
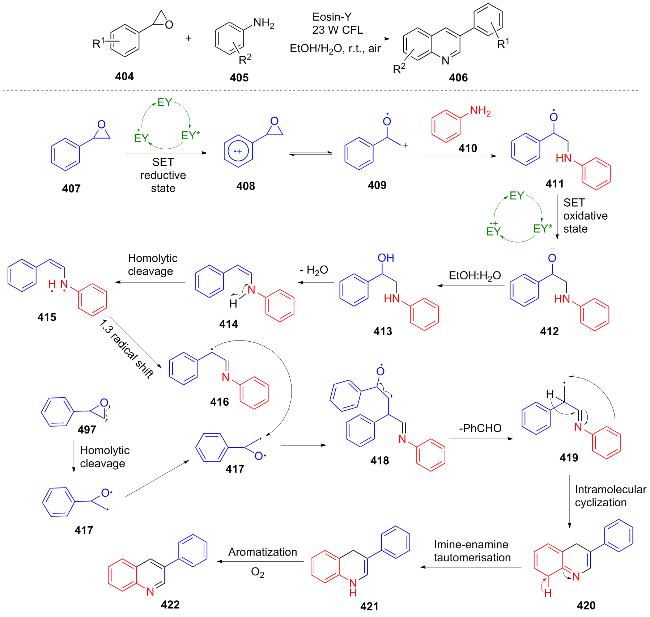
In the same year, Singh[87] achieved the transformation of 3-substituted indole 423 to C-3 aminoquinolin-2(1H)-one 423 through photocatalytic denitrogenation. They proposed a reasonable mechanism: TMSN3 was activated by 4-dimethylaminopyridine (DMAP) and H2O to generate $\text{N}_{3}^{}$, which underwent [3+2] cycloaddition with the substrate 423 to form a triazoline intermediate. The intermediate was oxidized by photoexcited Ir(III) to form a radical cation, which was deprotonated by DMAP to generate a radical. Subsequently, the radical underwent ring expansion and nitrogen gas release followed by rearrangement, and was finally reduced by the reduced state of the iridium catalyst, protonated and enolized to give the product (Scheme 45). This reaction achieves photocatalytically driven skeletal rearrangement, thereby avoiding the harsh reaction conditions typically associated with conventional nitration-reduction and Hofmann rearrangement processes. Notably, the reaction demonstrates a yield of 75% at the gram scale and is applicable to the synthesis of pharmaceutical molecules, such as HSP90 protein inhibitors.
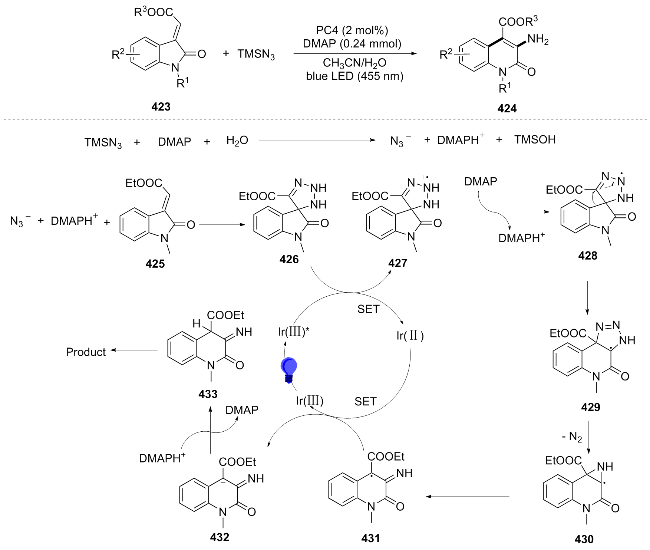
The semi-heterogeneous photo-redox catalytic system is an advanced catalytic strategy that combines the high efficiency of homogeneous catalysis with the easy separation of heterogeneous catalysis. In 2024, Li[88] used propargyl aniline derivatives 434, aryl diazonium tetrafluoroborates 435, and K2S2O5 as raw materials to achieve a radical cascade cyclization reaction under visible light through a semi-heterogeneous photo-redox catalytic system, which enabled the multi-component construction of C—S and C—C bonds and was used to synthesize biologically active 3-sulfonylquinoline derivatives 436. Under visible light, semiconductors generate photogenerated electrons and holes. The electrons reduce aryl diazonium salts to form phenyl radicals, which react with K2S2O5 to produce sulfonyl radicals 438. 438 adds to the substrate to form allyl radicals 439, which undergo intramolecular cyclization to form 440. The photogenerated holes oxidize the radicals generated from N-hydroxyphthalimide (NHPI), which in turn oxidize 440 to form dihydroquinoline 441. The latter then undergoes aromatization to produce the target product. NHPI is regenerated through the oxidation of 440 (Scheme 46). This reaction demonstrates broad substrate applicability and has been successfully scaled up to the gram level (yield 78%). Additionally, the catalyst recovery has been experimentally validated, showing that it can be recycled up to seven times without significant loss of catalytic activity.
In 2025, Wang et al.[89] synthesized a series of 2-(3- cyanoalkyl) substituted quinoline derivatives through the 6-endo-trig radical cyclization of cyclobutanone oximes and 1-isocyano-2-vinylbenzenes under the action of a photocatalyst. The excited state IrIII* reduced cyclobutanone oximes 445, generating imine radical 447 and IrIV through single-electron transfer. Radical 447 underwent C—C bond cleavage to form γ-cyano radical 448, which successively underwent radical addition with isocyanide and alkene to form carbon radical 449. Carbon radical 449 was oxidized by IrIV to form carbocation 451 (regenerating IrIII), and finally, the product 452 was obtained through deprotonation by base (Scheme 47). This study provided a modular synthetic route for 2-cyanoalkyl quinolines, but the cost of the catalyst and the applicability of some substrates (low yield for strongly electron-withdrawing groups or heterocyclic substrates) need to be improved.
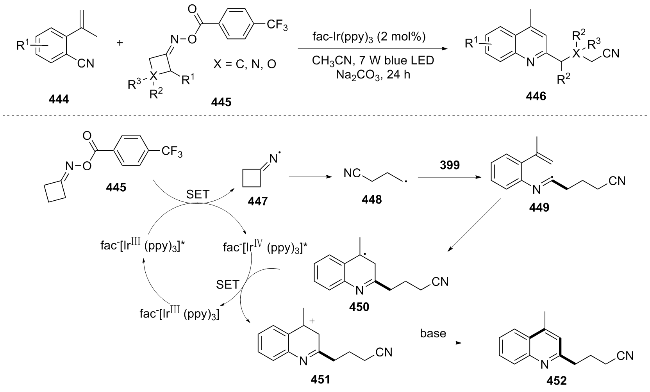
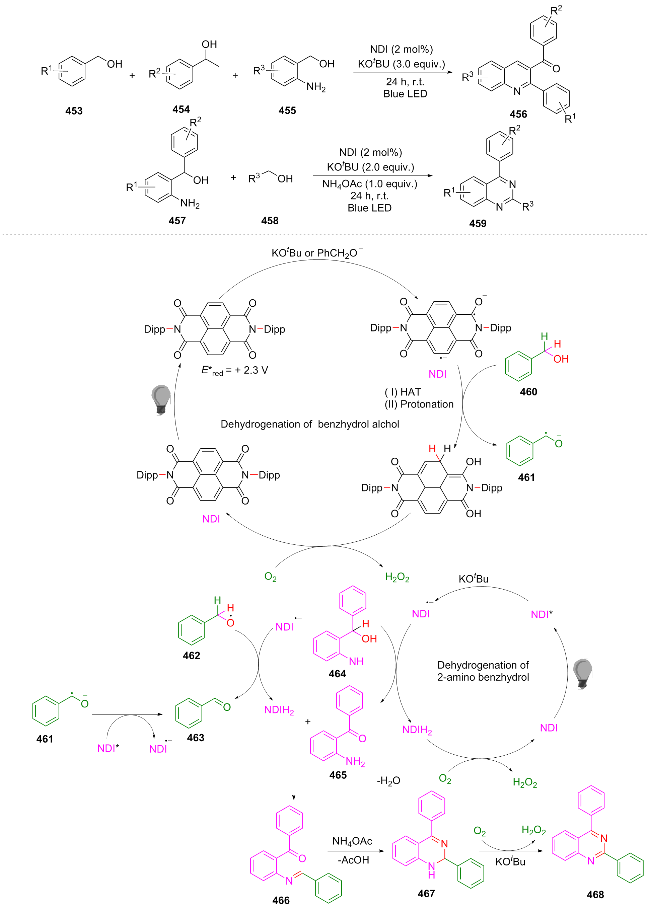
In the same year, Mandal et al.[90] utilized the highly reducing organic photocatalyst naphthalenediimine (NDI) to efficiently synthesize multi-substituted quinazolines and quinolines under ambient temperature and aerobic conditions. The core process of this photocatalytic cycle is as follows: the excited state NDI is reduced to the stable radical NDI⁻, which abstracts a hydrogen atom from benzyl alcohol to form NDIH2 and generate a benzoyl radical. This radical can either reduce another excited state NDI to form an aldehyde or transfer an electron to oxygen to form a superoxide radical. Oxygen then oxidizes NDIH2 to regenerate the NDI catalyst (releasing H2O2), driving the cycle. Compounds 460 and 464 are respectively oxidized to 463 and 465, and then undergo condensation, ammonium salt treatment and dehydrogenation, ultimately cyclizing to the quinazoline product 468 (Scheme 48). This methodology has enabled the successful synthesis of 32 quinazoline derivatives and six quinoline derivatives, with the gram-scale reaction proving to be feasible (yield 54%). It offers a mild and efficient synthetic approach for the preparation of quinazolines and quinolines. However, further optimization remains possible with respect to substrate scope expansion and reduction of base dependency.
Photocatalytic synthesis of quinoline compounds offers notable advantages over traditional methods. Conventional approaches typically require stoichiometric oxidants, leading to low atom economy and significant environmental impact, whereas photocatalysis eliminates the need for such reagents. Various photocatalytic systems exhibit distinct characteristics: most systems demonstrate broad substrate compatibility, particularly for substrates containing donor or acceptor groups, heterocycles, and aryl substituents (e.g., CuCl, PQ, dual catalytic systems). However, the applicability to aliphatic substrates (e.g., in the PQ system), substrates with strong electron-withdrawing groups, or specific heterocycles (such as in cyanoalkyl quinoline synthesis) remains limited. Organic dyes (e.g., Eosin Y, PQ, NDI) are cost-effective, operate under mild conditions, and exhibit high efficiency, yet they are often substrate-dependent. Precious metal catalysts (e.g., Ru and Ir complexes) demonstrate high catalytic activity but are associated with high costs. Semi-heterogeneous systems combine high efficiency with recyclability, although the design and optimization of multi-component systems can be complex. Mechanistically, photocatalysis primarily proceeds through single-electron transfer (e.g., Cu/PQ), energy transfer (e.g., PQ), or radical cascade pathways (e.g., semi-heterogeneous systems), thereby circumventing the use of conventional strong oxidants. Nevertheless, certain reactions still depend on molecular oxygen or additives. Although photocatalysis has achieved significant progress in sustainability, challenges persist, including the high cost of precious metals, substrate limitations, and system complexity. The development of a broadly applicable and recyclable photocatalytic platform represents a promising direction for future research.
8 Light-driven catalytic synthesis of qui- nazolines and its derivatives
Quinazolines represent a significant class of nitrogen-containing heterocyclic compounds that are commonly found in natural products and pharmaceuticals.[91] These compounds exhibit a diverse range of biological activities, including antimicrobial,[92] antimalarial[93] and antiviral[94] effects. Notably, quinazolines serve as key intermediates for several important therapeutic agents, such as alfuzosin (an adrenergic blocker), quinazoline (a muscle relaxant), and erlotinib (an anticancer agent) (Figure 8). Given the critical applications of the quinazoline backbone, there is an urgent need for the development of efficient and environmentally friendly synthetic methods.
The traditional synthetic strategies involve the reaction of ortho-functionalized anilines and their derivatives with aldehydes catalyzed by transition metals. These methods have drawbacks, including the need for expensive catalysts, ligands or additives, as well as harsh reaction conditions. Therefore, it is crucial to develop an efficient and environmentally friendly synthetic method that can be carried out under mild conditions.
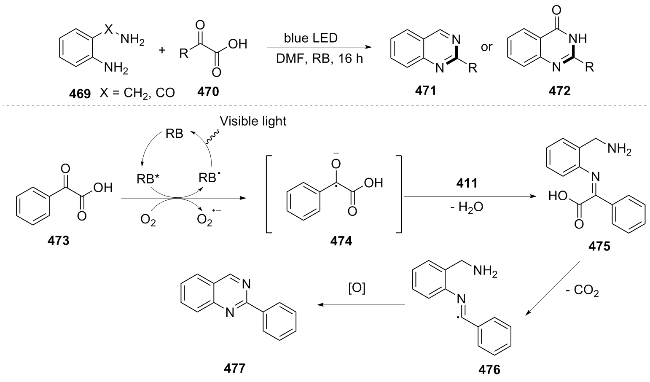
In 2022, Yang et al.[95] utilized Rose Bengal as a photosensitizer and, under room temperature and blue LED irradiation, efficiently constructed the quinazoline skeleton through a tandem condensation/decarboxylation reaction of 2-amino-benzylamine 469 and α-keto acid 470. A possible reaction mechanism was proposed: 473 reacts with the excited state rhodamine B (RB) under light to form the radical anion intermediate 474, which dehydrates to generate intermediate 475, and 475 further decarboxylates to form the radical intermediate 476, which then undergoes oxidation and cyclization to yield the product 477 (Scheme 49). This reaction overcame the limitations of traditional aldehyde substrates and provided a new strategy for the green synthesis of quinazoline drugs. However, the sub-strate scope (especially for aliphatic acids) and steric tolerance still need to be optimized.
In 2023, Li et al.[96] efficiently synthesized quinazoline derivatives using 2-aminobenzylamine 478 and methyl benzoylformate 479 as substrates. The reaction proceeds through two pathways: In pathway A, 482 is hydrolyzed by candida antarctica lipase B (CALB) to phenylglyoxylic acid, which is then oxidized under photocatalysis to form 484. 484 reacts with 485 to dehydrate and form 486, which is decarboxylated to 487 and oxidized to 488. In pathway B, 482 is photo-oxidized to 489, which condenses and dehydrates with 485 to form 490. 490 is hydrolyzed by CALB to 486, which is decarboxylated to 487 and oxidized to 488 (Scheme 50). This method is the first to combine lipase with organic photocatalysts, expanding the application of photo-enzyme co-catalysis in the synthesis of N-heterocycles. However, the low yield of aliphatic substrates remains a challenge that needs to be overcome.
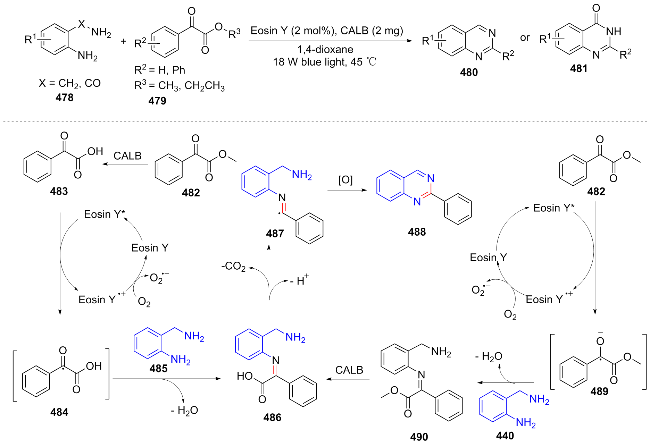
In the same year, Xie et al.[97] developed a novel visible light catalytic α-hydroxy acid decarboxylation strategy to achieve one-pot synthesis of quinazoline. PC oxidizes 492 to 492+•, reduces O₂ to $\text{O}{{_{2}^{}}^{-\ \bullet }}$ (PC regeneration). 492+• combines with $\text{O}{{_{2}^{}}^{-\ \bullet }}$ to form 494 and HOO•, 494 decarboxylates to 495. 495 reacts with HOO· and releases H₂O to form 496, 496 dehydrates and cyclizes with 497 to form 498, which is then oxidized by PC to 499, and finally 501 is obtained through the action of reactive oxygen species (Scheme 51). This study presents notable advantages, including the absence of metal catalysts, utilization of air oxidation, and a one-pot operational procedure. However, further optimization is required to address the substrate's steric sensitivity and the solvent's specificity.
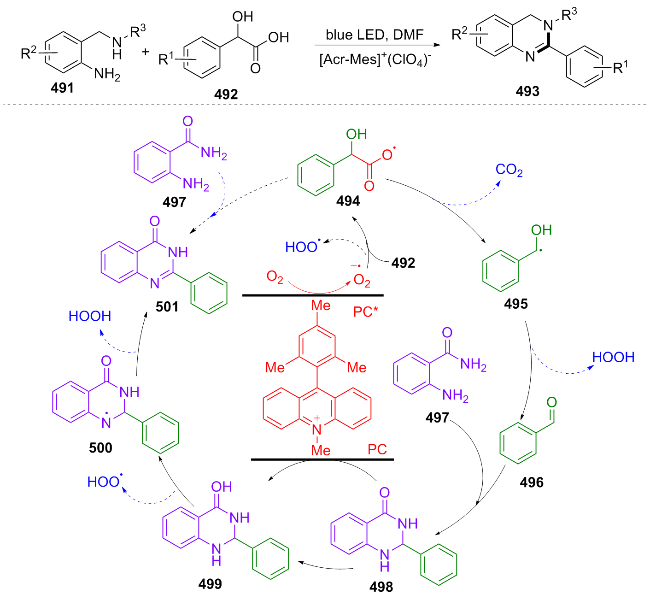
During that very year, Ma et al.[98] synthesized quinazoline compounds 502 using 2-aminoaryl ketone 503, aldehydes 504 and ammonium acetate as raw materials. They proposed that the reaction mechanism was as follows: 2-aminoaryl ketone 505 and aldehyde 506 underwent nucleophilic addition dehydration under blue LED light to form intermediate 507. Then, the carbonyl group of intermediate 507 reacted with ammonium acetate to generate ketimine 508, which underwent intramolecular cyclization to form dihydroquinazoline 509, and finally aromatized to produce the product 510 (Scheme 52). This study presents an efficient and environmentally benign catalytic-free approach for the synthesis of quinazolines through a one-pot three-component reaction strategy. The methodology de- monstrates potential applications in pharmaceutical chemistry and the synthesis of functional materials. However, it exhibits sensitivity to steric hindrance, as evidenced by the relatively low yields observed with ortho-substituted aromatic aldehydes.
In 2024, Yang et al.[99] developed a novel method for the synthesis of 2-substituted quinazoline derivatives 513 through the synergistic catalysis of visible light and organic selenium. They proposed a possible mechanism for this reaction: (PhSe)2 dissociates under blue light to form PhSe• and PhSe+, PhSe• undergoes single-electron transfer with O2 to generate PhSe+ and $\text{O}{{_{2}^{}}^{-\ \bullet }}$, while benzylamine 514 is photoexcited to 515, which is dehydrogenated to imine 516 under the action of PhSe+/PhSe•. Then, 516 couples with another benzylamine and deaminates to 517, which is hydrolyzed to benzaldehyde 518 and then dehydrates and condenses with 2-aminobenzylamine 519 to yield 520 (Scheme 53). This method provides a new approach for organic selenium catalysis and green drug synthesis.
In the same year, Arumugam et al.[100] used o-acylani- lines, trialkylamines and NH4Cl as raw materials, Eosin Y as a photocatalyst, and under blue LED irradiation to synthesize 2,4-disubstituted quinazolines. Visible light excitation of EY generates EY*, which oxidizes triethylamine to form free radical 524 and is reduced to EY•⁻. EY•⁻ is oxidized by O2 to regenerate the catalyst and generate $\text{O}{{_{2}^{}}^{-\ \bullet }}$. $\text{O}{{_{2}^{}}^{-\ \bullet }}$ abstracts a proton from 524 to form imine intermediate 525 and HOO. 525 hydrolyzes to give key aldehyde 527, while substrate 521 condenses with NH4Cl to form 528. 528 condenses with 527 to form imine bond 529, which undergoes 6-endocyclic ring closure and photo-oxi- dative aromatization to give quinazoline product (Scheme 54). This method has wide substrate universality. Notably, this method can synthesize medicinal molecules (such as midazolam impurity H), expanding the application of tertiary amines in C—N bond activation and providing new ideas for the construction of drug molecules.
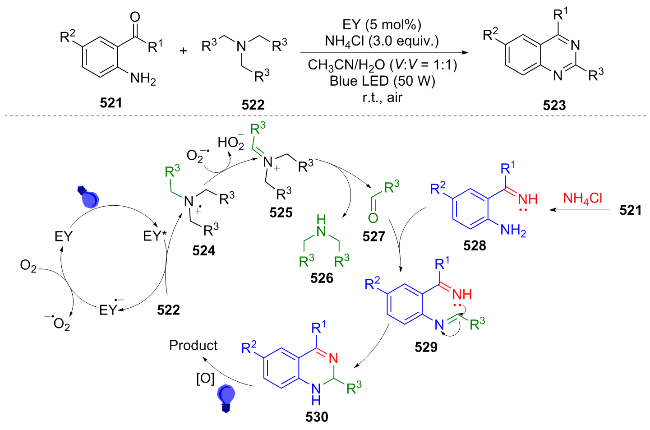
In 2025, Aazath et al.[101] developed a novel photocatalyst based on lignin-derived carbon-capped CuO nanocomposites (CuO/C) for the efficient synthesis of quinazoline compounds 533 under visible light irradiation. The authors proposed a reasonable mechanism: photoexcitation of CuO/C generates OH· and $\text{O}{{_{2}^{}}^{-\ \bullet }}$, and $\text{O}{{_{2}^{}}^{-\ \bullet }}$ oxidizes NH4OAc to form an amine radical. This radical condenses with the ketone group of 531 to form 534, and its amino group condenses with 532 to form 535, which then cyclizes and desorbs from the catalyst as 536 (Scheme 55). This methodology demonstrates high product yields (84%~94%), broad substrate applicability encompassing various functional groups such as halogen, methoxy, and nitro substituents, and magnetic recoverability of the catalyst, which can be reused for five cycles while retaining 90% its initial activity.
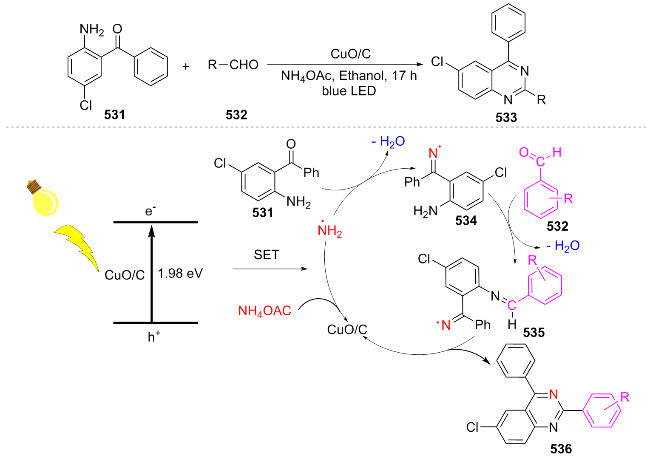
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Compared with traditional synthetic routes that rely on transition metal catalysts, strong oxidants, and harsh conditions, photocatalytic methods offer notable advantages. By employing a visible light-driven, oxygen-mediated radi- cal mechanism, these methods enable the environmentally benign construction of quinazoline skeletons at room temperature, thereby avoiding metal contamination and high energy consumption. Different photocatalytic systems demonstrate distinct characteristics in terms of application scope and performance. For instance, Rose Bengal, Eosin Y, and organic selenium-based catalytic systems exhibit excellent compatibility with aromatic substrates containing halogen, alkoxy, and other functional groups (yield>80%). However, their effectiveness is generally limited when applied to aliphatic substrates such as aliphatic aldehydes and acids (yield<50%). The catalyst-free three-component approach can accommodate certain aliphatic aldehydes, whereas the enzyme-photocatalytic synergistic system shows considerable yield variability with heterocyclic substrates. Future research should focus on overcoming current limitations, including poor reactivity toward aliphatic substrates, steric sensitivity, and challenges related to scale-up.
9 Summary and outlook
The photocatalytic synthesis of nitrogen-containing heterocyclic compounds through a visible light-driven radical mechanism has significantly overcome the reliance of traditional methods on high-temperature conditions, precious metal catalysts, and stoichiometric oxidants. This approach enables green and highly atom-economical transformation processes under mild reaction conditions. The numerous examples listed in this review fully demonstrate the broad application prospects of such methods in organic synthesis, drug development, and materials science. With the popularization of cost-effective light sources, the development of photocatalytically active materials, and the increasing social focus on environmentally sustainable technologies, photochemical methods have been significantly promoted. Moreover, existing studies have shown that various photocatalytic systems can achieve gram-scale synthesis and have been successfully applied in the construction of drug molecules, thereby significantly enhancing their practical application value.
However, this technology still faces several challenges during its development. For instance, its applicability to aliphatic substrates is limited, the cost of noble metal catalysts is high, and there are significant differences in reaction efficiency in complex systems. To achieve further breakthroughs, future research should focus on developing new catalysts with broader substrate applicability, optimizing catalyst preparation processes to reduce the reliance on noble metals, and actively exploring reaction systems without the need for photocatalysts. At the same time, promoting gram-scale and even larger-scale synthesis is of great significance for the practical industrial application of this technology. Particularly in the pharmaceutical field, solving the problem of potential contamination of the final product by metal ions in photocatalysts is especially crucial. The ultimate goal should be to achieve a smooth transition from laboratory-scale green synthesis to large-scale industrial pharmaceutical production.
(Lu, Y.)