

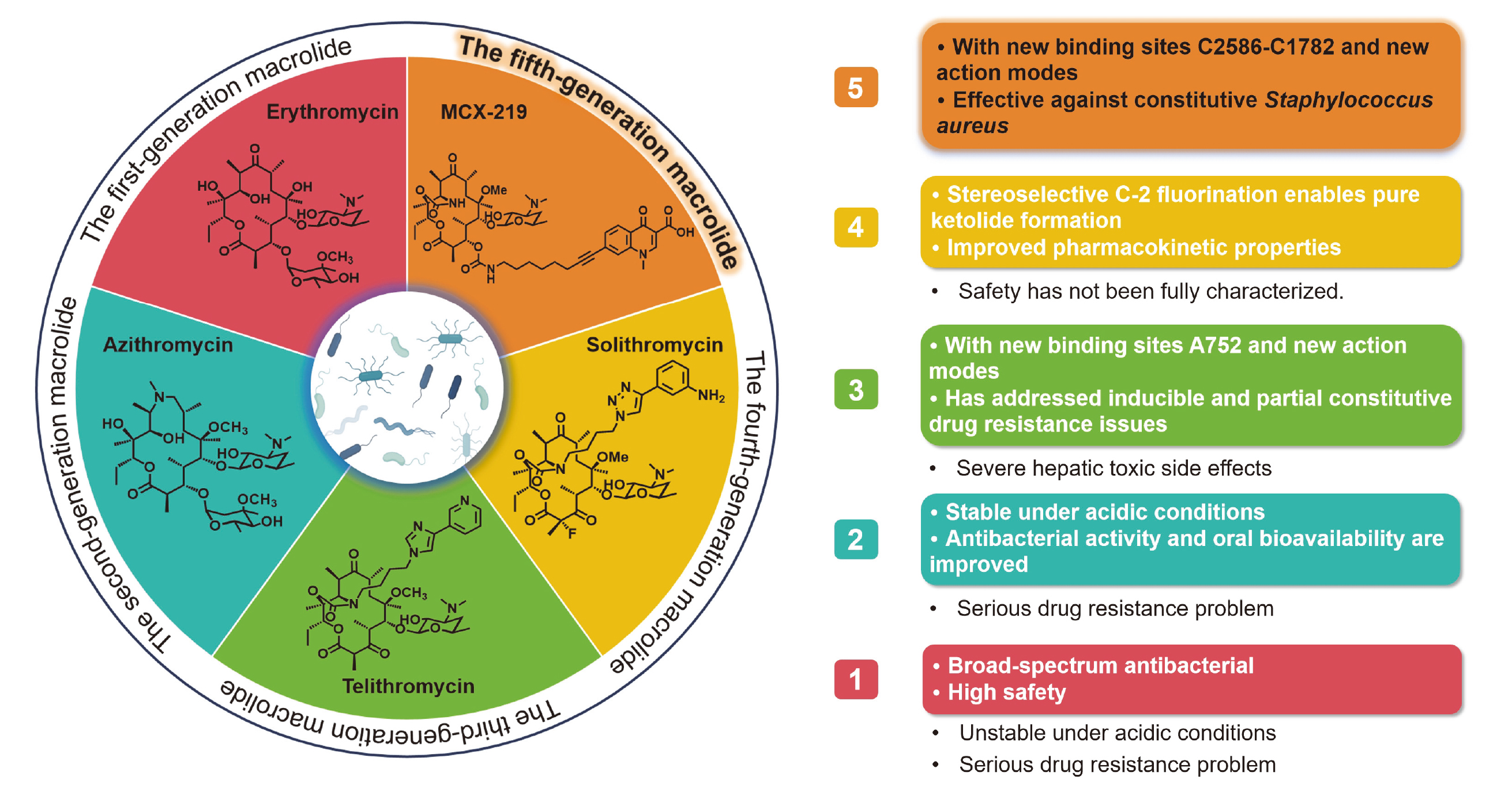
1 大环内酯的发展
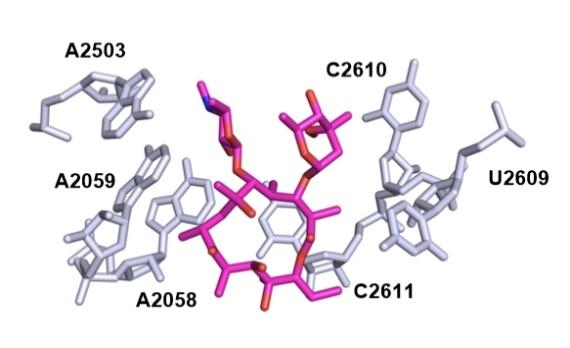
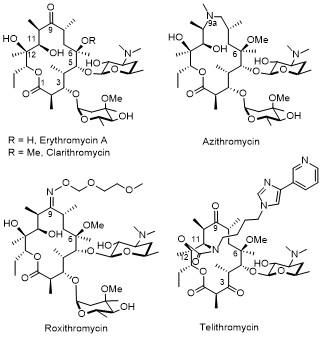
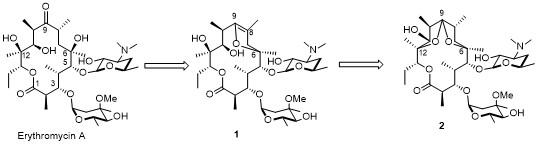
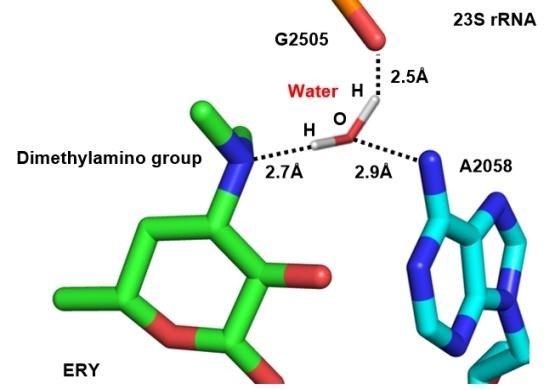
表1 2013~2024年大环内酯与70S核糖体复合物结构PDB汇总Table 1 Summary of PDB structures of macrolide-70S ribosome complexes from 2013 to 2024 |
| Macrolide | Species | Res/Å | Method | PDB No. | Ref. |
|---|---|---|---|---|---|
| Erythromycin | E. coli | 3.60 | Electron microscopy | 5JU8 | [25] |
| Erythromycin | E. coli | 3.60 | Electron microscopy | 5JTE | [25] |
| Telithromycin | B. subtilis | 3.10 | Electron microscopy | 6HA1 | [26] |
| Telithromycin | E. coli | 3.50 | Electron microscopy | 6HA8 | [26] |
| Erythromycin | Thermus thermophilus | 2.85 | X-Ray diffraction | 6ND6 | [27] |
| Erythromycin | Staphylococcus aureus | 2.42 | Electron microscopy | 6S0X | [28] |
| Erythromycin | Thermus thermophilus | 2.55 | X-Ray diffraction | 6XHX | [8] |
| Telithromycin | Thermus thermophilus | 2.60 | X-Ray diffraction | 6XHY | [8] |
| Erythromycin | E. coli | 2.90 | Electron microscopy | 7NSO | [29] |
| Erythromycin | E. coli | 3.50 | Electron microscopy | 7NSP | [29] |
| Telithromycin | E. coli | 3.10 | Electron microscopy | 7NSQ | [29] |
| Erythromycin | E. coli | 3.00 | Electron microscopy | 7Q4K | [30] |
| Erythromycin | Thermus thermophilus | 2.50 | X-Ray diffraction | 8FC1 | [31] |
| Erythromycin | Thermus thermophilus | 2.45 | X-Ray diffraction | 8FC4 | [31] |
| Azithromycin | Thermus thermophilus | 2.50 | X-Ray diffraction | 8FC2 | [31] |
| Azithromycin | Thermus thermophilus | 2.65 | X-Ray diffraction | 8FC5 | [31] |
| Telithromycin | Thermus thermophilus | 2.35 | X-Ray diffraction | 8FC6 | [31] |
| Telithromycin | Thermus thermophilus | 2.60 | X-Ray diffraction | 8FC3 | [31] |
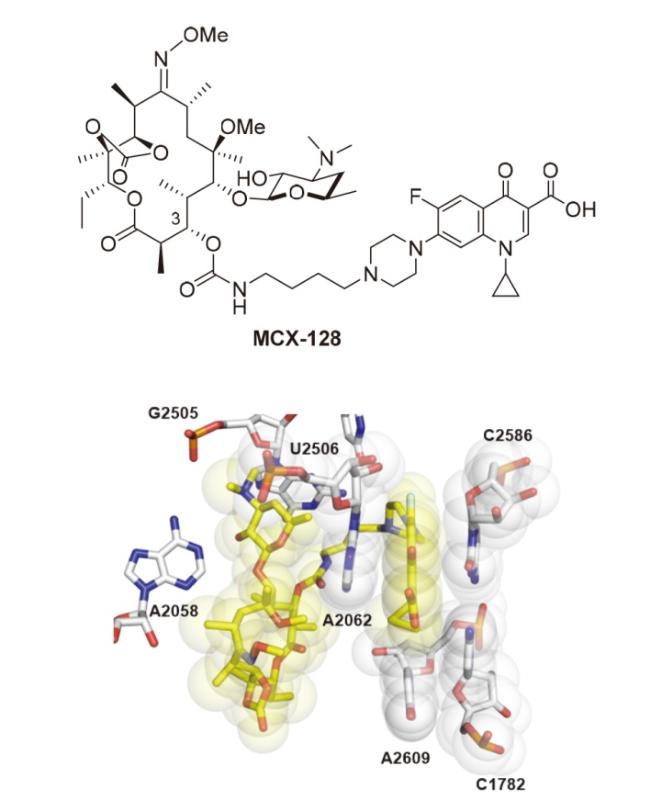
| MCX-66 | Thermus thermophilus | 2.40 | X-Ray diffraction | 8VTU | [32] |
| MCX-128 | Thermus thermophilus | 2.40 | X-Ray diffraction | 8VTX | [32] |
| MCX-128 | Thermus thermophilus | 2.35 | X-Ray diffraction | 8VTW | [32] |
| MCX-91 | Thermus thermophilus | 2.40 | X-Ray diffraction | 8VTV | [32] |
| MCX-190 | Staphylococcus aureus | 2.58 | Electron microscopy | 8Y38 | [23] |
| MCX-190 | Staphylococcus aureus | 3.60 | Electron microscopy | 8Y39 | [23] |
表2 2013~2024年大环内酯与50S核糖体复合物结构PDB汇总Table 2 Summary of PDB Structures of Macrolide-50S Ribosome Complexes from 2013 to 2024 |
| Macrolide | Species | Res/Å | Method | PDB No. | Ref. |
|---|---|---|---|---|---|
| Carbamolide 4d | Deinococcus radiodurans | 3.20 | X-Ray diffraction | 4IO9 | [33] |
| Carbamolide 4e | Deinococcus radiodurans | 3.20 | X-Ray diffraction | 4IOA | [33] |
| Carbamolide 4f | Deinococcus radiodurans | 3.60 | X-Ray diffraction | 4IOC | [33] |
| Erythromycin | E. coli | 6.60 | Cryo-electron microscopy | 3J5L | [34] |
| Erythromycin | E. coli | 3.90 | Cryo-electron microscopy | 3J7Z | [35] |
| Telithromycin | Staphylococcus aureus | 3.43 | X-Ray diffraction | 4WF9 | [36] |
| Erythromycin | Deinococcus radiodurans | 3.54 | X-Ray diffraction | 4WFN | [37] |
| Erythromycin | Staphylococcus aureus | 2.30 | Electron microscopy | 6S0Z | [28] |
| Clarithromycin | Mycobacterium tuberculosis | 3.30 | Electron microscopy | 7F0D | [38] |
| Erythromycin | Mycobacterium smegmatis | 3.60 | Electron microscopy | 8XZ3 | [39] |
| MCX-190 | Staphylococcus aureus | 2.65 | CElectron microscopy | 8Y36 | [23] |
| MCX-190 | Staphylococcus aureus | 2.53 | Electron microscopy | 8Y37 | [23] |
2 第三代大环内酯类抗生素
2.1 酮内酯(Ketolide)
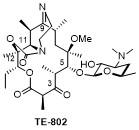
2.1.1 TE-802
2.1.2 泰利霉素
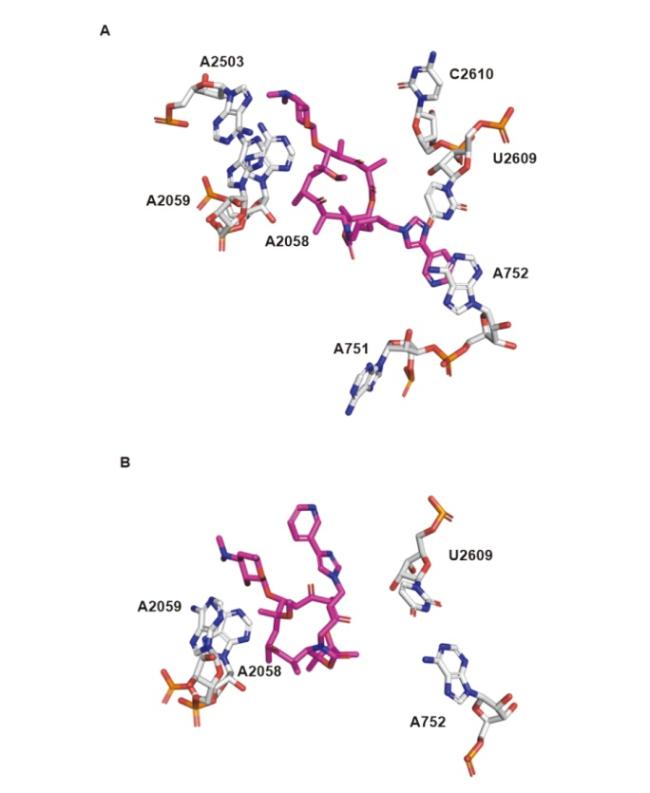
图5 (A)泰利霉素与嗜热栖热菌核糖体靶点A752的相互作用[粉红色结构为泰利霉素(PDB: 4V7Z)][51]; (B)泰利霉素与金黄色葡萄球菌核糖体的复合物结构[泰利霉素在金黄色葡萄球菌中与A752不具有相互作用, 粉红色结构为泰利霉素(PDB: 4WF9)][36]Figure 5 (A) Interaction between telithromycin and ribosomal target A752 of Thermus thermophilus (The pink structure is telithromycin (PDB: 4V7Z));[51] (B) Structure of the complex between telithromycin and ribosomes of S. aureus (Telithromycin does not interact with A752 in S. aureus. The pink structure is telithromycin (PDB: 4WF9))[36] |
2.1.3 新型酮内酯
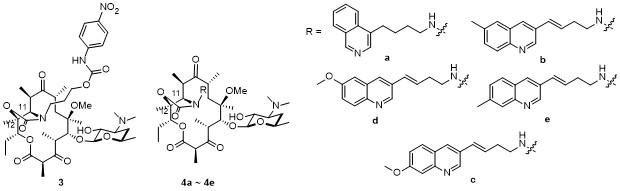
2.1.3.1 11位修饰酮内酯
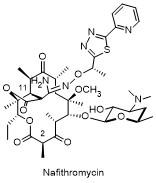
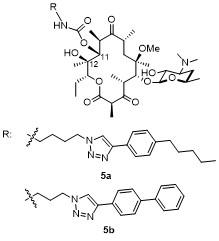
2.1.3.2 9位修饰酮内酯
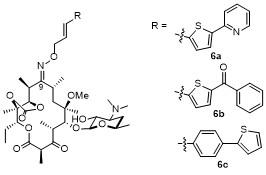
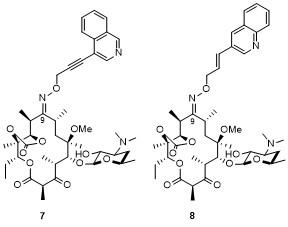
2.1.3.3 德胺糖修饰酮内酯
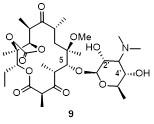
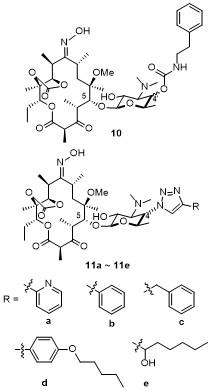
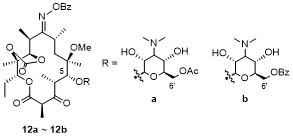
2.1.3.4 其余位点修饰酮内酯
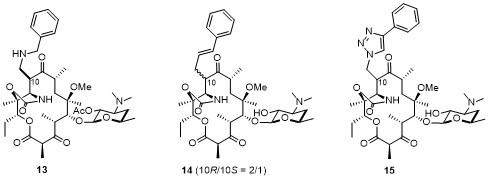
表3 化合物14的部分抗菌活性Table 3 Antibacterial activity of compound 14 |
| Strain | Resistance phenotypes (genotypes) | MIC/(μg•mL-1) | |
|---|---|---|---|
| 14 | Clarithromycin | ||
| Streptococcus pneumoniae BAA1402 | M (mef) | 0.03 | 2 |
| Staphylococcus aureus BAA976 | M (mef) | 0.03 | 64 |
| Staphylococcus aureus BA977 | iMLS (erm) | 0.03 | >64 |
2.2 非酮内酯
2.2.1 酰内酯(Acylide)
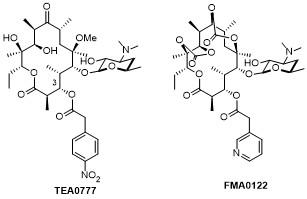
2.2.1.1 C-11,12为碳酸酯的酰内酯
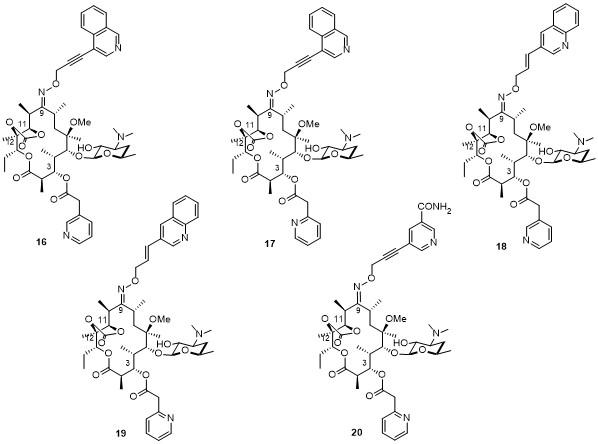
2.2.1.2 C-11,12为氨基甲酸酯的酰内酯
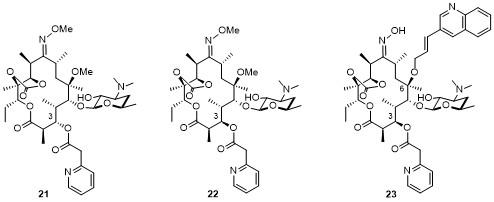
2.2.1.3 C-11,12为羟基的酰内酯
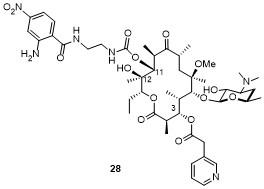
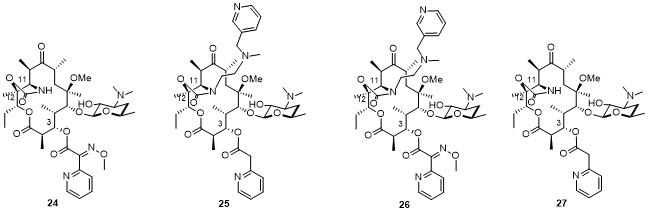
2.2.2 氨基甲酸内酯(Carbamolide)
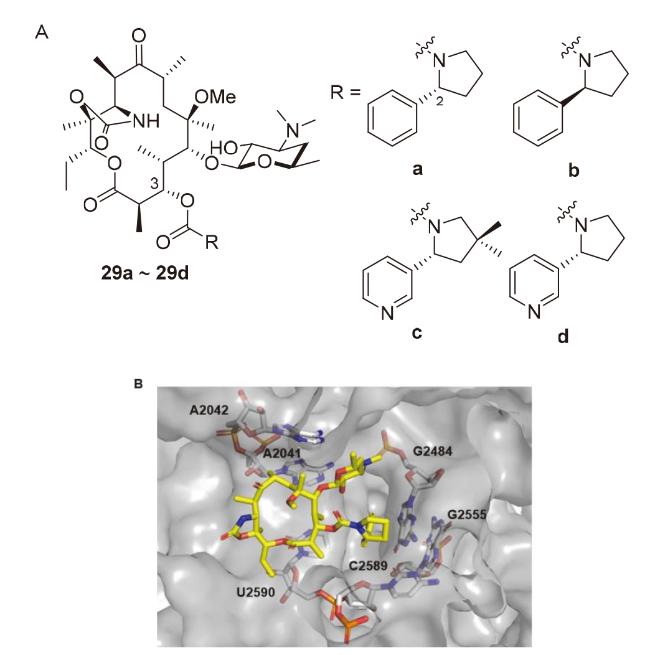
表4 化合物29a和29b的部分抗菌活性Table 4 Antibacterial activity of compounds 29a and 29b |
| Strain | Resistance phenotypes | MIC/(mg•L-1) | ||
|---|---|---|---|---|
| 29a | 29b | Clarithromycin | ||
| Streptococcus pneumoniae 1243-00 | MLSB | 0.5 | 64 | >64 |
| Streptococcus pyogenes 1304-00 | MLSB | 0.5 | 32 | >64 |
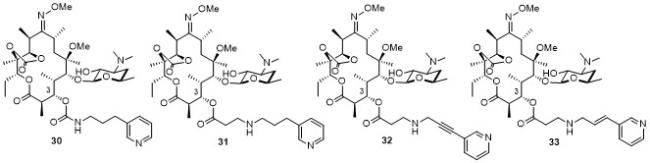
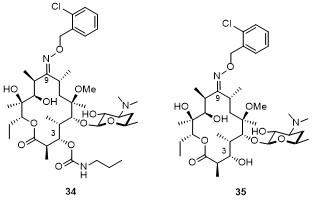
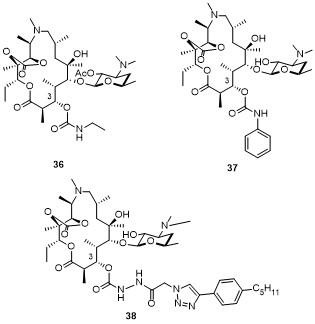
2.2.3 烃基内酯(Alkylide)
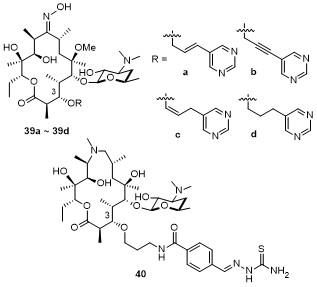
2.2.4 非酮内酯的糖基修饰
2.2.4.1 非酮内酯克拉定糖修饰
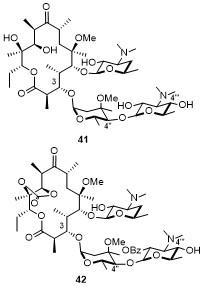
表6 化合物41和42的部分抗菌活性Table 6 Antibacterial activity of compounds 41 and 42 |
| Strain | Resistance phenotypes | MIC/(μg•mL-1) | |||
|---|---|---|---|---|---|
| 41 | 42 | Telithromycin | Clarithromycin | ||
| Staphylococcus epidermidis 12-4 | MRSEa | 4 | 8 | ≤2 | 32 |
| Staphylococcus epidermidis 12-11 | MRSE | 2 | ≤1 | ≤2 | 4 |
a MRSE: Methicillin-Resistant Staphylococcus epidermidis |
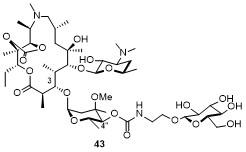
表7 化合物43的部分抗菌活性Table 7 Antibacterial activity of compound 43 |
| Strain | Resistance phenotypes | MIC/(μg•mL-1) | |
|---|---|---|---|
| 43 | Azithromycin | ||
| Streptococcus pneumoniae 943 | Sensitive | 1 | 4 |
| Streptococcus pneumoniae 746 | Sensitive | 2 | 8 |
| MRSA-1 | MRSAa | 16 | 16 |
a MRSA: Methicillin-Resistant Staphylococcus aureus |
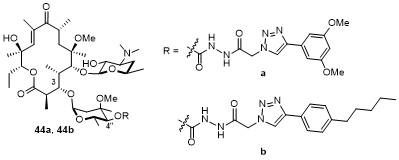
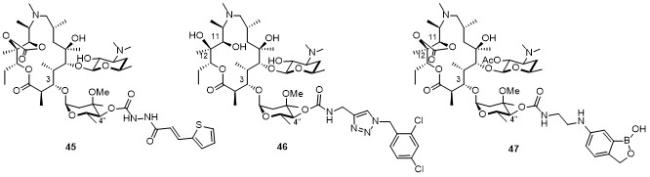
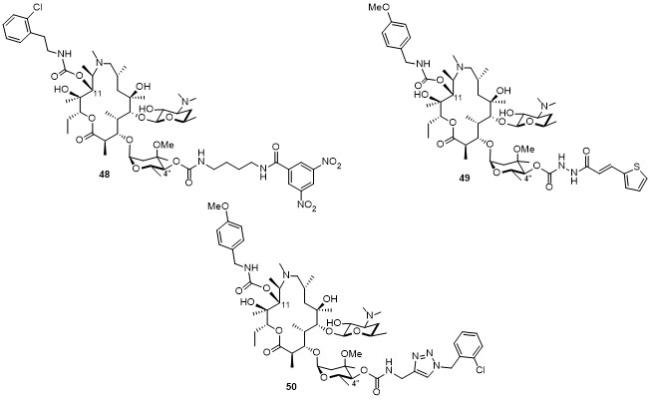
表8 化合物48的部分抗菌活性Table 8 Antibacterial activity of compound 48 |
| Strain | Resistance genotypes | MIC/(μg•mL-1) | |
|---|---|---|---|
| 48 | Azithromycin | ||
| Streptococcus pneumoniae B1 | erm | 0.25 | 128 |
| Streptococcus pneumoniae AB11 | mef, erm | 2 | 256 |
| Streptococcus pneumoniae A22072 | mef | 0.03 | 4 |
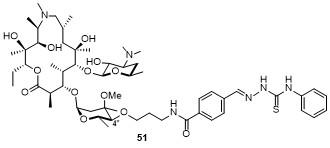
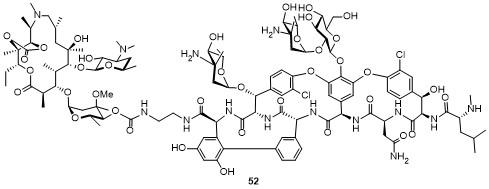
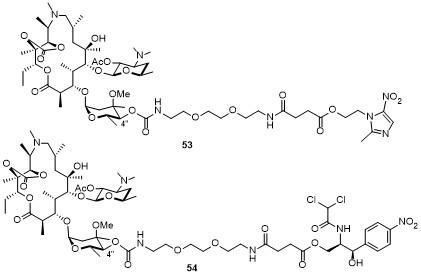
2.2.4.2 非酮内酯德胺糖修饰
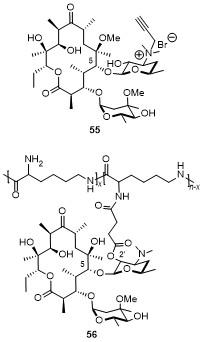
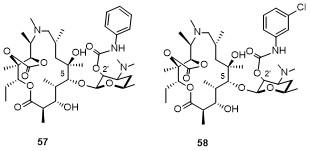
3 第四代大环内酯类抗生素
3.1 索利霉素
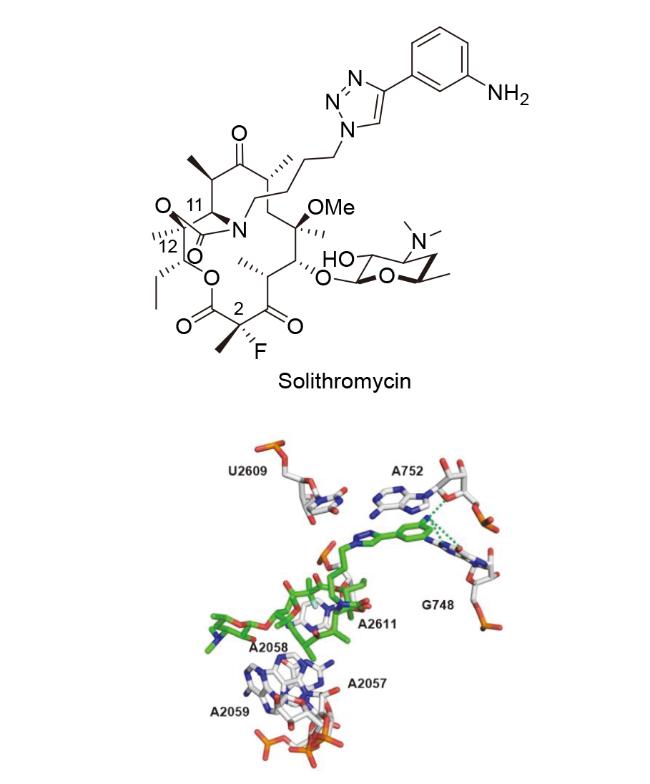
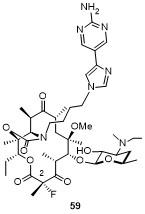
3.2 新型第四代大环内酯抗生素
3.2.1 引入联芳基的侧链
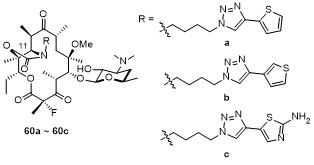
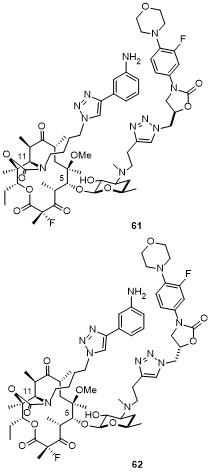
3.2.2 引入稠合芳基的侧链
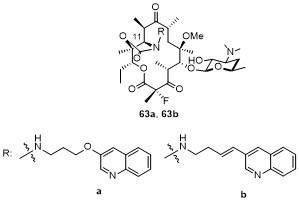
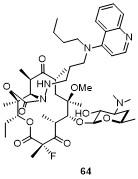
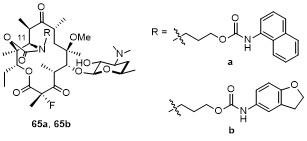
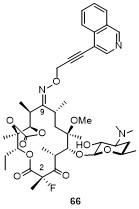
表9 化合物66的抗菌活性Table 9 Antibacterial activity of compound 66 |
| Strain | Resistant determinants | MIC/(μg•mL-1) | |
|---|---|---|---|
| 66 | Telithromycin | ||
| Streptococcus pneumoniae PU09 | PSSP,a mef | 0.25 | 0.25 |
| Streptococcus pyogenes 01-968 | i-erm | 0.062 | 0.062 |
| Streptococcus pyogenes 12-207 | mef | 0.031 | 0.5 |
a PSSP: Penicillin-Susceptible Streptococcus pneumoniae |
4 非天然大环内酯类抗生素全合成
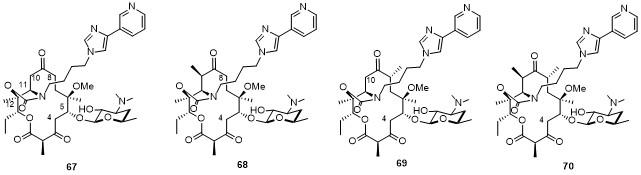
4.1 去甲基化泰利霉素全合成
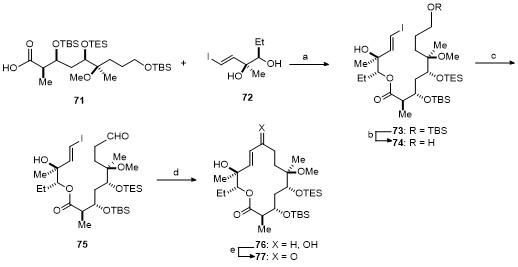
图式2 分子内Nozaki-Hiyama-Kishi反应完成环化Scheme 2 Intramolecular Nozaki-Hiyama-Kishi reaction completes cyclization Reagents and conditions: (a) Cl3PhCOCl, Et3N, DMAP, 75% yield; (b) TBAF, AcOH, 60% yield; (c) DMP, NaHCO3, 78% yield; (d) CrCl2, cat. NiCl2, DMSO, 50% yield; (e) DMP, 80% yield. |
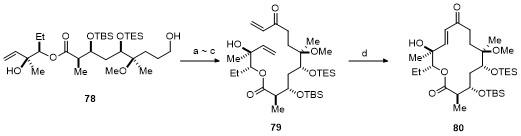
图式3 分子内环化复分解反应完成环化Scheme 3 Intramolecular ring-closing metathesis reaction completes cyclization Reagents and conditions: (a) Dess-Martin Periodinane, NaHCO3; (b) vinyl MgBr, THF; (c) Dess-Martin periodinane, CH2Cl2, 60% yield over three steps; (d) 20 mol% Grubbs II cat., 60% yield. |
表10 去甲基泰利霉素的抗菌活性Table 10 Antibacterial activity of demethyltelithromycin |
| Strain | Resistant determinant | MIC/(μg•mL-1) | ||||
|---|---|---|---|---|---|---|
| 67 | 68 | 69 | 70 | Telithromycin | ||
| E. coli SQ171/2058G | A2058G | >512 | >256 | >256 | >256 | — |
| E. coli DK/PKK3535 | wt | 32 | 4 | 8 | 0.5 | 0.5 |
| E. coliDK/2058G | A2058G | 64 | 32 | 16 | 4 | 1 |
| Staphylococcus aureus UCN14 | A2058T | 32 | >256 | >256 | >256 | >128 |
| Staphylococcus aureus ATCC33591 | ermA | >128 | >64 | >128 | >128 | >128 |
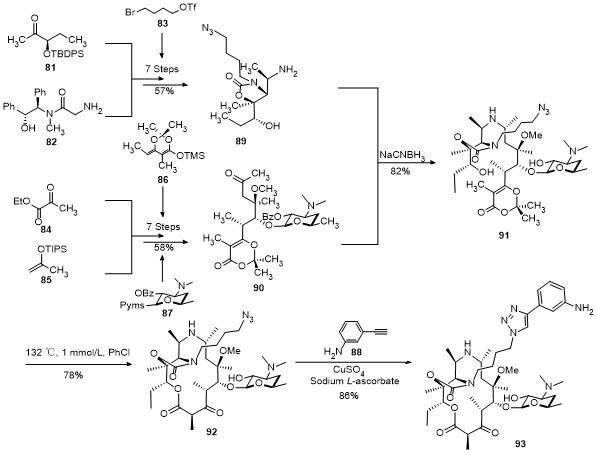
4.2 氮杂酮内酯和索利霉素全合成
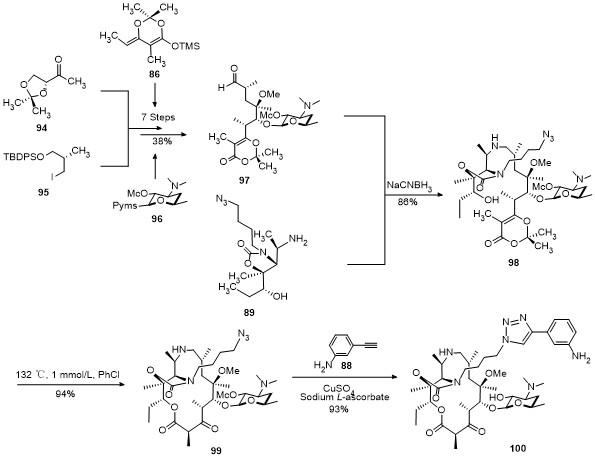
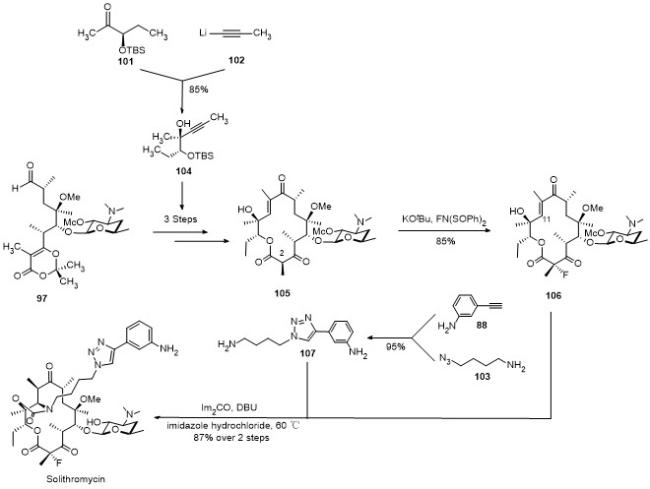
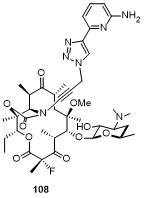
5 大环内酯-喹诺酮杂合物(Macrolones)
5.1 C-4''位连接喹诺酮
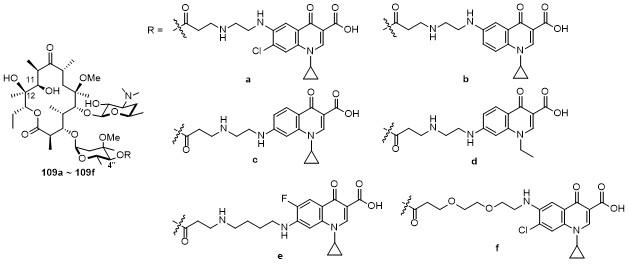
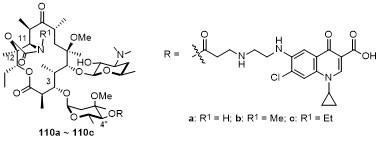
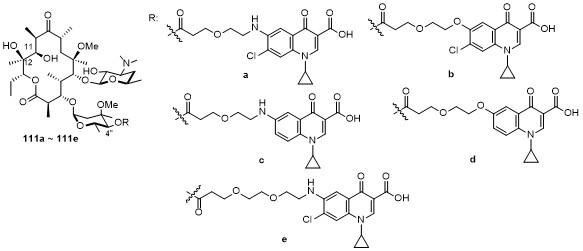
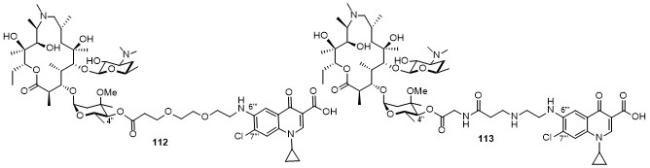
5.2 C-6位连接喹诺酮
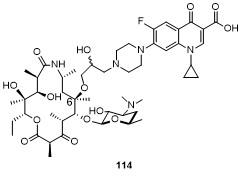
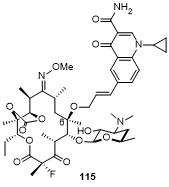
表11 化合物115的部分抗菌活性Table 11 Antibacterial activity of compound 115 |
| Strain | Resistant determinant | MIC/(μg•mL-1) | |||
|---|---|---|---|---|---|
| 115 | Azithromycin | Telithromycin | Ciprofloxacin | ||
| Streptococcus pneumoniae 07P390 | c-ermB | ≤0.008 | 256 | 0.016 | — |
| Streptococcus pyogenes 12-206 | c-ermTR | 0.016 | >256 | 0.031 | 0.125 |
| Staphylococcus aureus PU32 | MRSA, i-ermA | 0.25 | 32 | 0.125 | 64 |
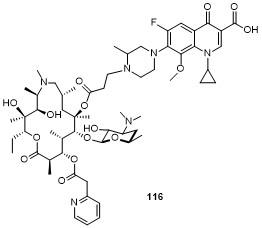
5.3 C-3位连接喹诺酮
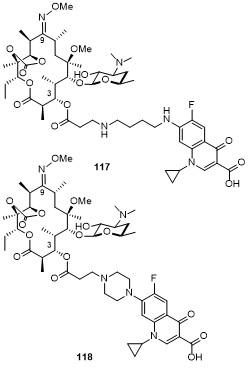
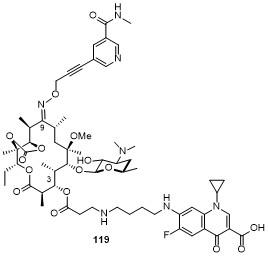
表12 化合物119的部分抗菌活性Table 12 Antibacterial activity of compound 119 |
| Strain | Resistant determinants | MIC/(μg•mL-1) | |
|---|---|---|---|
| 119 | Telithromycin | ||
| Streptococcus pneumoniae PU09 | mef | ≤0.008 | 0.5 |
| Streptococcus pyogenes 12-206 | c-ermTR | 0.06 | 0.25 |
| Haemophilus influenzae ATCC49247 | sensitive | 2 | 4 |
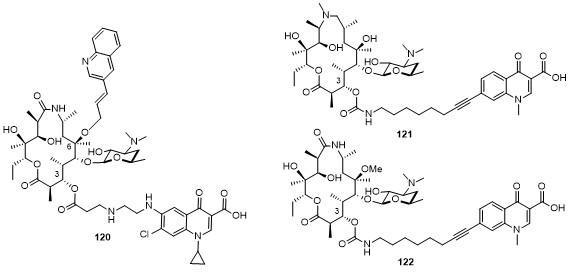
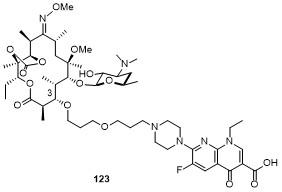
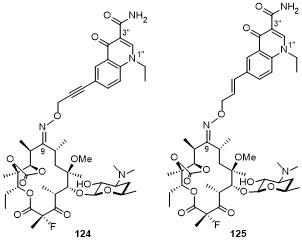
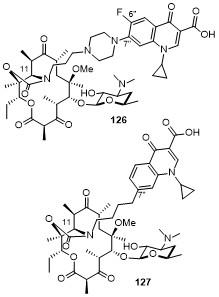
5.4 C-9/C-11位连接喹诺酮
6 第五代大环内酯类抗生素
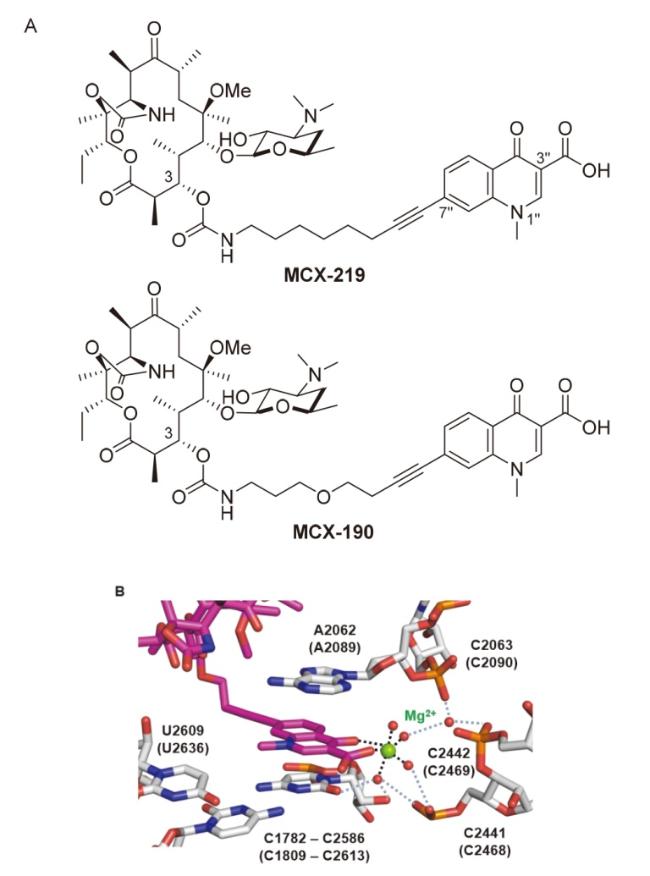
图58 (A)化合物MCX-219和190的结构及(B)化合物MCX-190与靶点的新作用模式[括号中是金黄色葡萄球菌rRNA编号, 粉色结构为化合物MCX-190 (PDB: 8Y36)][23]Figure 58 (A) Structures of compounds MCX-219 and 190, and (B) new binding mode of compound MCX-190 with the target [The rRNA number of S. aureus is in parentheses. The pink structure represents compound MCX-190 (PDB: 8Y36)][23] |
表13 化合物MCX-190和MCX-219的部分抗菌活性Table 13 Antibacterial activity of compounds MCX-190 and MCX-190 |
| Strain | Resistant determinants | MIC/(μg•mL-1) | ||
|---|---|---|---|---|
| MCX-190 | MCX-219 | Telithromycin | ||
| Staphylococcus aureus 15B196 | c-ermB | 8 | 1 | 32~>256 |
| Staphylococcus aureus PU32 | i-ermA | 0.25~0.5 | 0.25 | 0.12~0.5 |
| Mycoplasma pneumoniae | A2058G | 1 | 0.25 | 256 |
| Mycoplasma pneumoniae | A2059G | 2 | 0.25 | 256 |
| Mycoplasma pneumoniae | A2058T | 0.5 | 0.06 | 256 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}