

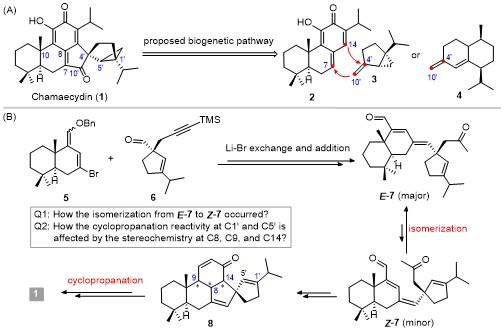
1 Introduction
Since 1983, nine cryptoquinonemethides including eight C30-terpenes and one C35-terpene were isolated from the plant genera of Cryptomeria.[1-5] All the members of this family share an unprecedented spiro[4.4]nonane-containing 6-6-6-5-5-3 hexacyclic skeleton (e.g. 1 in Scheme 1A). In addition, they have five to seven chiral centers including two or three all-carbon quaternary ones at the points of C4'-spirocenter and C10 and/or C1' ring fusion. The proposed biogenetic pathway for the formation of such a unique polycyclic ring system is arisen from double linkage at C7—C10' and C4'—C14 between an abietane-type diterpenoid (2) and a thujane-type monoterpenoid (3) for the C30-terpene,[2] or a cadinane-type sesquiterpenoid (4) for the C35-terpene.[5] The C4'-stereo- isomer of chamaecydin 1, known as isochamaecydin, should be generated via the C4'—C10' bond rotation during the C4'—C14 bond forming reaction.
The biological property of these natural products has not been extensively evaluated presumably due to the scarcity of natural sources. However, it was reported that chamae- cydin 1 exhibited significant antifeedant activity at 20 μg/disk against the pest insect Spodoptera litura.[4] In addition, the C35-terpene known as cryptotrione showed an IC50 value of (6.44±2.23) μmol/L against the human oral epidermoid carcinoma KB cells,[5] which is only slightly weaker than the clinically used anticancer drug etoposide (VP-16, IC50=2.0 μmol/L). The data suggest that the cryptoquinonemethides should have widespread and promising biological activities.
Owing to the intriguing structural features and promising biological activity, the synthesis of cryptoquinone- methides has garnered the great interests from the synthetic community in recent years. The first synthesis of the member in cryptoquinonemethide family was reported by the group of Wong/Peng in 2020,[6] who achieved an elegant synthesis of C35-terpene cryptotrione in its racemic form. The key transformations for the construction of polycyclic ring system involved a Rh-catalyzed C—H insertion,[7] a Pt-catalyzed 1,5-enyne cycloisomerization,[8] and a Bi(OTf)3-mediated polyene cyclization.[9] In 2023, Xie and coworkers[10] reported a nice synthesis of (-)- chamaecydin (1) and its C4'-isomer (-)-isochamaecydin, which are the corresponding enantiomers of naturally occurring (+)-chamaecydin and (+)-isochamaecydin, from the “chiral pool” terpenes. In this synthesis, an Ireland- Claisen rearrangement,[11] a Rh-catalyzed C—H insertion,[7] and a Bi(OTf)3-promoted polyene cyclization[9] were employed to build the ring skeleton. Very recently, we[12] accomplished the first catalytic asymmetric synthesis of (+)-chamaecydin and (+)-isochamaecydin, as well as their C1'—C5' stereoisomers via a modular approach. In addition to these total syntheses, two synthetic studies for efficient construction of the polycyclic frameworks of cryptotrione have also been reported by the group of Shi[13] and Li/He/Hu.[14]
As illustrated in Scheme 1B, for the synthesis of (+)-chamaecydin and its 1',5'-epimer, a convergent route for the construction of the 6-6-6-5-5 pentacyclic system (8) has been developed, through the Li-Br exchange/addition reaction of vinyl bromide 5 with aldehyde 6, followed by a base-promoted isomerization of E-7 to Z-7 and a concomitant intramolecular Michael/aldol cascade or Robinson annulation of Z-7. Subsequently, the C13-carbonyl group directed regio- and stereo-selective cyclopropanation[15-18] at C1'=C5' double bond of pentacycle 8 furnished the construction of [3.1.0]-bicyclic system of 1. During our synthetic study, the Li-Br exchange/addition reaction of 5 with 6 afforded a mixture of E-7 and Z-7. However, the E-isomer was produced predominantly with an E/Z ratio of ca. 8∶1. This raises a question on how the E-7 is isomerized to Z-7 during the Michael/aldol cyclization. On the other hand, the reactivity as well as the regio- and stereo-selectivity for the cyclopropanation of 8 was markedly affected by the stereochemistry at C9, C8, and C14. This observation poses another question of how the variations in configurations impact the cyclopropanation reactivity. To address these issues, we recently carried out detailed investigations to gain an in-depth understanding of the reasons. The detailed results will be presented herein.
2 Results and discussion
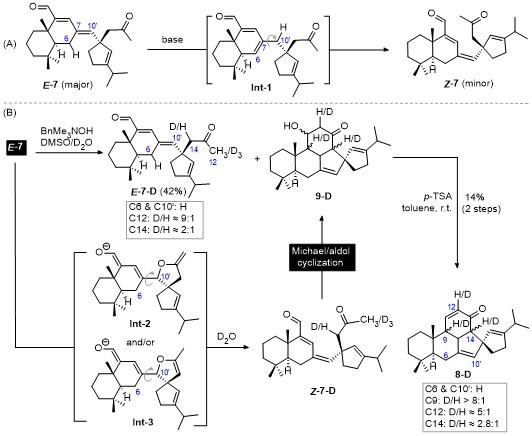
2.1 Investigation into the mechanism of the isomerization of E-7 to Z-7
In our initial design of the synthetic route, we envisioned that, based on the related report,[19] the isomerization of thermodynamically more stable E-7 to less stable Z-7 might proceed via the rotation of C7—C10' single bond in Int-1 (Scheme 2A), which might be formed through the prototropic shift between C6 and C10' associated with the double bond shift from C7=C10' to C6=C7. Interestingly, however, control experiments by adding a small amount of D2O in the reaction system revealed that the deuteration was not occurred at C6 and/or C10' as seen from 1H NMR spectra of the recovered starting material E-7-D (Scheme 2B). Instead, C12 and C14 were deuterated significantly with a D/H ratio of ca. 9∶1 and 2∶1, respectively, as determined by 1H-NMR analysis. On the other hand, dehydration of the Michael/aldol product 9-D afforded 8-D. In consistent with the observation of recovered E-7-D, 1H NMR analysis revealed that deuteration at C6 and/or C10' in 8-D was also not observed. Whereas C12 and C14 were deuterated with a D/H ratio of ca. 2.8∶1 and 5∶1, respectively. In addition, significant deuteration at C9, which should occur during the Michael/aldol cyclization, was also observed, although the relatively precise D/H ratio could not be calculated out due to a severe overlap of H9-proton with others. These overall results strongly support that, differing from our initially considered prototropic shift process via Int-1, the isomerization of E-7 to Z-7 should proceed reversibly through oxa-1,6- addition and retro-oxa-1,6-addition process via Int-2 and/or Int-3.
2.2 Investigation into the cyclopropanation reactivity of the pentacyclic system
As shown in Scheme 3, the base-promoted isomerization and concomitant intramolecular Michael/aldol cyclization of the E/Z mixture 7 produced pentacyclic 8a/b as a couple of diastereoisomers at C9, C8, and C14.[12] It was found that the cyclopropanation of the mixture proceeded regioselectively at C1'=C5' double bond to give the cyclopropanated product 10 in 33% yield accompanied by the recovery of starting material in 53% yield. Interestingly, NMR analyses revealed that both the product 10 and the recovered starting material were obtained as a single diastereoisomer. Thus, based on the yields of product and recovered starting material, as well as their NMR data, we could conclude that the minor isomer 8b could react smoothly to give product 10. In contrast, the major isomer 8a was almost unreactive under the employed reaction conditions. These results indicate that the cyclopropanation reactivity of the two diastereoisomers of 8 is largely different. Based on the 1H NMR, 13C NMR, 1H-1H COSY, HSQC, HMBC, and 1H-1H NOESY spectroscopic analyses of 10, a correlation between H9 & H20 could be observed, indicative of H9β configuration. However, the absolute configuration at C8 and C14 as well as the stereoselectivity of cyclopropane could not be determined due to the significant overlap of proton signals. Fortunately, by transforming 10 to 11 through a two-step procedure involving the addition of C13-carbonyl group with iPrLi followed by pyrindium chlorochromate (PCC) oxidation,[20-22] the correlation relationships between H9 & H20, H8 & H14, H14 & H5', and H15 & H6' in compound 11 could be clearly observed through multiple NMR spectroscopic analyses. These correlations affirmed that the stereochemistries of H9, H8, and H14 for 11 should be H9β, H8α, and H14α, respectively. In addition, the reaction should proceed stereoselectively at β-face presumably due to the directing- effect of the carbonyl group at C13. Thus, the absolute configuration at C9, C8, and C14 for the more reactive diastereoisomer of 8b could be deduced to be H9β, H8α, H14α.
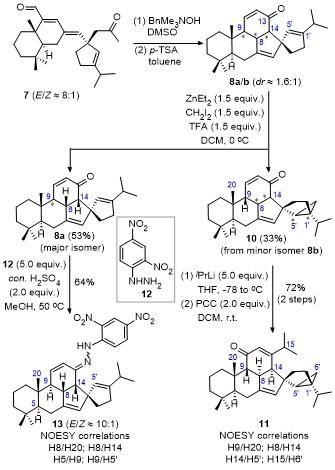
On the other hand, for the less reactive diastereoisomer 8a, while a strong correlation between H20 and H8/H14 could be observed, it was difficult to confirm both H8 and H14, or only one of them, correlated with H20 due to the complete overlap of H8 and H14. In addition, the exact configuration of H9 was also difficult to be assigned due to severe overlap of H9 with other protons. After extensive trials, the hydrazone 13, derived from 8a and hydrazine 12, was a suitable derivative.[23] Multiple NMR spectroscopic analyses on 13 revealed that the stereochemistries at C9, C8, and C14 were H9α, H8β, and H14β according to the observed correlations between H8 & H20, H8 & H14, H5 & H9, and H5' & H9. The results, in turn, affirm that the configurations at C9, C8, and C14 for 8a are H9α, H8β, and H14β, which are opposite to the corresponding diastereoisomer 8b.
The pentacyclic compound 15 is the C4'-epimer of 8, which was constructed from 14,[12] the C4'-epimer of 7, as a mixture of diastereoisomers 15a/b with a dr ratio of ca. 1.6∶1 (Scheme 4A). Similar to 8a/b, it was also found that the two diastereoisomers exhibited different reactivity for cyclopropanation reaction since the cyclopropanated product 16 and the recovered starting material 15b were obtained as a single diastereoisomer. However, an entire assignment of the absolute configurations at C8, C9, and C14 for both 16 and 15b, and the stereochemistry of cyclopropane in 16 was problematic based on their NMR spectral information. Accordingly, 16 was converted into 17, the key intermediate for accessing 1',5'-epi-isochamae- cydin,[12] according to the same procedures for the synthesis of 11. Fortunately, a combination of multiple NMR spectroscopic analyses of 1H NMR, 13C NMR, 1H-1H COSY, HSQC, HMBC, and 1H-1H NOESY of 17 affirmed that the absolute conformations at C9, C8, and C14 were H9α, H8β, and H14β, respectively, and the cyclopropane group at C1'—C5' was directed toward β-face. On the other hand, while the absolute configuration of 15b was correctly proposed in our previous study,[12] sufficient supports did not been obtained at the time. The problems were, although the absolute configuration at C9 could be determined as H9β based on the observation of clear interactions between H9 and H20 as well as H9 and H5' from the spectra of both 15b, the assignment of the absolute configurations at C8 and C14 were somewhat difficult because of severe overlap of H8 and H14 with other protons. Whereas the relative stereochemistry of H8 and H14 could be confirmed to be cis-configuration. Accordingly, based on the experiences on the configuration assignment of 13, 15b was converted into its hydrazone 18. However, the assignment of the absolute configurations at C8 and C14 for 18 was still problematic owing to significant overlap of proton signals similar as for 15b.
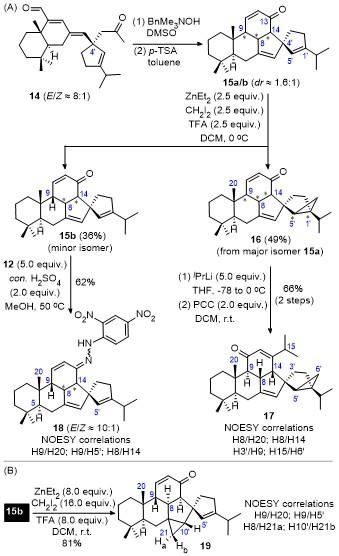
On the basis of the experiences that the cyclopropanated derivatives, such as 11 in Scheme 3 and 17 in Scheme 4, could provide useful spectroscopic information for the determination of the stereochemistry, the cyclopropanation of 15b under relatively harsher conditions had been carried out by using a large excess amount of cyclopropanation reagents and elevating the reaction temperature (Scheme 4B). Unexpectedly, structural characterization on the reaction product by multiple NMR spectroscopic analyses demonstrated that the reaction occurred regioselectively at C7=C10' position with α-face stereoselectivity, giving 19 as a single isomer in high yield. The cyclopropanated pro- duct at C1'=C5' double bond was not observed. In addition, 1H-1H NOESY spectrum showed clear correlations between H9 & H20, H9 & H5', and H8 & H21a, respectively. These observations, together with the cis-configu- ration of H8 and H14 as confirmed from 15b and 18, uneventfully demonstrated that the absolute configurations at C9, C8, and C14 for 19 are H9β, H8α, and H14α, respectively. As such, the absolute stereochemistries of H9, H8, and H14 for the less reactive 15b could be determined to be H9β, H8α, and H14α.
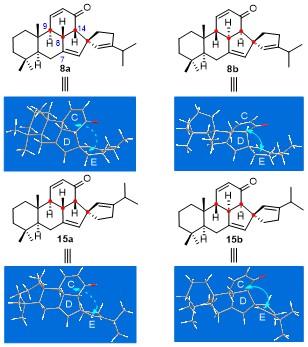
Thus, through a patient and thorough investigation, the stereochemical permutations at C9, C8, and C14 of all four diastereoisomers 8a/b and 15a/b could been unambiguously determined. As shown in Figure 1, the absolute configurations of H9, H8, and H14 are H9β, H8α, and H14α for 8a; H9β, H8α, and H14α for 8b; H9α, H8β, and H14βfor 15a; and H9β, H8α, and H14α for 15b, respectively. Among which, the cyclopropanation of 8b and 15a could readily proceed at C1'=C5' double bond with β-face stereoselectivity relative E-ring. In stark contrast, 8a and 15b were much less reactive. To have an in-depth understanding for how the variations in stereochemistry influence the reactivity, the structures were optimized by using density functional theory (DFT) calculation (see Figure 1). Based on the 3D structures of the four compounds, the different cyclopropanation reactivity of each diastereoisomer in response to the variation of the stereochemistries at C9, C8, and C14. Namely, the cyclohexenone ring (C-ring) in 8a twists significantly towards back right; as a result, the C1'=C5' double bond locating at the back side of E-ring is shielded by C-ring, thereby, displaying low reactivity. In contrast, the C-ring in 8b tilts toward front right, making the space around the back-side C1'=C5' double bond more opened, and consequently, exhibiting a higher reactivity.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The reactivity of the diastereoisomers 15a and 15b, which are the C4'-epimers of 8a and 8b, with the C1'=C5' double bond positioned at the front side of E-ring, could also be reasonably explained according to the stereochemical variations at C9, C8, and C14. Namely, for 15a possessing the same absolute configurations at C9, C8, and C14 as 8a, the C-ring also twist to back right as 8a. Therefore, the space around the front-side C1'=C5' double bond is more opened, and consequently, the cyclopropanation of C1'=C5' double bond occurs smoothly. On the other hand, for 15b with absolute configurations at C9, C8, and C14 identical to 8b, the C-ring apparently falls toward front right side. Such an orientation increases the steric hindrance around C1'=C5' double bond, thereby decreasing its reactivity.
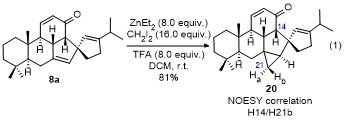
The theoretical calculation results also provide useful supports for explaining the outcome of the cyclopropanation of 15b. As clearly seen from the optimized 3D structure of 15b, the space at the α-face of central D-ring is much more opened than that at the β-face of D-ring and that of both α-/β-face of E-ring on the one hand, and the C7=C10' double bond should have a larger ring strain than the C1'=C5' double bond on the other hand. These overall factors result in the cyclopropanation of 15b proceeds regio- and stereo-selectively at the C7=C10' double bond with α-face selectivity relative to D-ring, affording 19 in high yield (Scheme 4). On the basis of these explanations, it may be predicted that the cyclopropanation of 8a should occur at the C7=C10' double bond with β-face stereoselectivity. As expected, our predication is unambiguously demonstrated by the experimental results (Eq. 1). That is, multiple NMR spectroscopic analyses for the product 20 obtained from the cyclopropanation of 8a[15-18] showed that the reaction proceeded uneventfully at the C7=C10' double bond with β-face selectivity of D-ring.
3 Conclusions
In summary, focusing on two questions regarding the isomerization mechanism of E- to Z-isomer and how the variation in stereochemistry impacts the cyclopropnation reactivity of a 6-6-6-5-5 pentacyclic system encountered during our synthetic studies on the total synthesis of cryptoquinonemethide natural products, a detailed investigation was carried out to address these issues. Through deuterium monitoring experiments, the mechanism for the isomerization of E- to Z-isomer proceeding via an oxo-1,6-addition and retro-oxo-1,6-addition process, unlike the reported prototropic shift. Subsequently, through a detailed multiple NMR spectroscopic analyses on different derivatives of the pentacyclic intermediates, the absolute configurations of all four diastereoisomers were clearly assigned. Finally, the 3D structures of the diastereoisomers were optimized by using DFT calculation based on the absolute configurations. On the basis of all these results, the variations in stereochemistries at C9, C8, and C14 cause the twist of central C-ring toward different directions, which results in different steric hindrance around C7=C10' and C1'=C5' double bonds, and therefore, showing different reactivity and selectivity. The results provide useful supports not only for explaining and predicting the different reactivity as well as stereo- and regio-selectivity for the cyclopropanation of the pentacyclic intermediates, but also for the design of other reactions and synthetic routes of relevant natural products.
4 Experimental section
4.1 General Information
Unless otherwise noted, all oxygen or moisture sensitive reactions were conducted in flame-dried glassware under nitrogen or argon atmosphere. Dry tetrahydrofuran (THF) and dichloromethane (DCM) were obtained from a solvent purification system on Innovative Technology. Other solvents and reagents were purchased from commercial sources and were used without further purification. An oil bath was used as heating source for reactions that require heating. Analytical thin layer chromatography (TLC) was performed on 0.2 mm thick silica gel 60-F254 plates (Merck) and visualized by exposure to ultraviolet light, or an ethanolic solution of phosphomolybdic acid. Column chromatographic purification of products was accomplished using forced-flow chromatography on 100~200 or 230~400 mesh silica gel. The 1H NMR spectra were recorded at 300, 400, or 500 MHz; and 13C NMR spectra were recorded at 75, 101, or 126 MHz on Bruker AV. Chemical shifts are given relative to TMS or the appropriate solvent peak. High resolution mass spectra (HRMS) were obtained on an IonSpec Ultima 7.0 T FT-ICR-MS (IonSpec, USA) using ESI or APCI as ionization method. Melting points were measured using a micro-melting point apparatus.
4.2 Deuterium labeling experiments for the isomerization of E-7 to Z-7
To a stirred solution of E-7 (38.3 mg, 0.100 mmol) in dimethyl sulfoxide (DMSO)/D2O (2 mL, V∶V=10∶1) was added BnMe3NOH [16.7 mg, 40% (w) in MeOH, 0.040 mmol, 0.4 equiv.] at room temperature under N2 atmosphere. The reaction mixture was stirred for 15 min at the same temperature and quenched by saturated aqueous NH4Cl. The solution was diluted with water (5 mL) and extracted with EtOAc (4 mL×4). The combined organic layers were washed successively with water (5 mL×3) and brine, then dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Purification of the residue by column chromatography on silica gel [eluent: V(petroleum ether)∶ V(EtOAc)=15∶1] afforded compound E-7-D (16.0 mg, 0.042 mmol, 42% yield) as a colorless oil and crude Michael/aldol product 9-D.
(4aS,8aS,E)-3-(((S)-3-Isopropyl-1-(2-oxopropyl)cyclo-pent-2-en-1-yl)methylene)-5,5,8a-trimethyl-3,4,4a,5,6,7,8,8a-octahydronaphthalene-1-carbaldehyde (E-7-D): 1H NMR (300 MHz, CDCl3) δ: 9.29 (s, 1H), 6.59 (s, 1H), 6.00 (d, J=2.2 Hz, 1H), 5.44~5.42 (m, 1H), 2.83 (d, J=15.7 Hz, 1H), 2.76~2.69 (m, 1.65H), 2.37~2.28 (m, 3H), 2.11 (s, 0.3H), 2.09~2.03 (m, 2H), 1.97~1.86 (m, 1H), 1.66~1.56 (m, 1H), 1.53~1.49 (m, 1H), 1.46~1.40 (m, 1H), 1.25~1.17 (m, 2H), 1.11 (s, 3H), 1.13~1.08 (m, 1H), 1.05 (d, J=6.7 Hz, 3H), 1.04 (d, J=6.5 Hz, 3H), 0.91 (s, 3H), 0.89 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 193.7, 152.3, 151.4, 148.3, 145.4 (×2), 136.6 (×2), 127.2, 51.8 (×2), 50.7, 41.9, 38.0 (broad), 37.3, 35.3, 33.6, 33.5, 32.0, 31.5, 29.9, 22.6, 21.6, 21.5, 21.4, 19.2, 18.8; HRMS (ESI) calcd for C26H36D3O2 [M+H]+ 386.3133, found 386.3129; calcd for C26H35D4O2 [M+H]+ 387.3196, found 387.3191; calcd for C26H34D5O2 [M+H]+ 388.3258, found 388.3249.
To a stirred solution of Michael/aldol product 9-D in toluene (1 mL) was added p-TSA (p-methylbenzene- sulfonic acid, 17.2 mg, 0.100 mmol, 1.0 equiv.) at room temperature. The reaction mixture was stirred for 3 h at room temperature and quenched by saturated aqueous NaHCO3. The resulting mixture was extracted with EtOAc (5 mL×3). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Purification of the residue by column chromatography on silica gel [eluent: V(petroleum ether)∶V(EtOAc)=20∶1] afforded compound 8-D (5 mg, 13.7 μmol, 14% yield over 2 steps, dr=ca. 3∶2) as a colorless oil.
(4R,6aS,10aR)-3'-Isopropyl-7,7,10a-trimethyl-3a,3a1,6,6a,7,8,9,10,10a,10b-decahydro-3H-spiro[acephenanthryl-ene-4,1'-cyclopentan]-2'-en-3-one (8-D): 1H NMR (300 MHz, CDCl3) δ: 7.05~7.02 (m, 0.6H, major), 6.91~6.88 (m, 0.4H, minor), 5.94 (d, J=10.2 Hz, 0.17H), 5.20~5.18 (m, 0.6H, major), 5.08 (s, 0.6H, major), 5.05 (s, 0.4H, minor), 4.81~4.79 (m, 0.4H, minor), 3.15~3.09 (m, 0.6H, major), 2.88~2.77 (m, 0.66H), 2.55~2.43 (m, 1H), 2.38~2.14 (m, 4H), 2.12~2.00 (m, 1.42H), 1.87~1.76 (m, 1.63H), 1.71~1.51 (m, 3H), 1.51~1.41 (m, 1H), 1.30~1.15 (m, 1H), 1.10~1.02 (m, 6H), 1.00~0.95 (m, 4H), 0.92~0.87 (m, 6H); 13C NMR (major isomer, 101 MHz, CDCl3) δ: 201.1, 151.8, 148.6, 148.5, 143.6, 131.3, 131.2, 130.7, 128.7, 65.7, 65.6, 54.0, 52.6, 49.7, 47.4 (×3), 41.8, 37.0, 35.9, 34.2, 34.1, 33.8, 33.0, 29.9, 25.3, 22.3, 22.0, 21.7, 21.6, 19.8; 13C NMR (minor isomer, 101 MHz, CDCl3) δ: 200.1, 152.9, 147.6, 147.5, 144.1, 132.8, 132.7, 128.2, 125.4, 65.1 (×2), 58.6, 56.7, 54.7, 48.4 (×3), 42.4, 39.3, 39.2, 37.8, 37.1, 34.0, 33.3, 31.9, 30.0, 25.9, 21.7, 21.6, 21.3, 18.7, 15.9; HRMS (ESI) calcd for C26H36DO [M+H]+ 366.2902, found 366.2898; HRMS (ESI) calcd for C26H35D2O [M+H]+ 367.2965, found 367.2956; HRMS (ESI) calcd for C26H34D3O [M+H]+ 368.3027, found 368.3010.
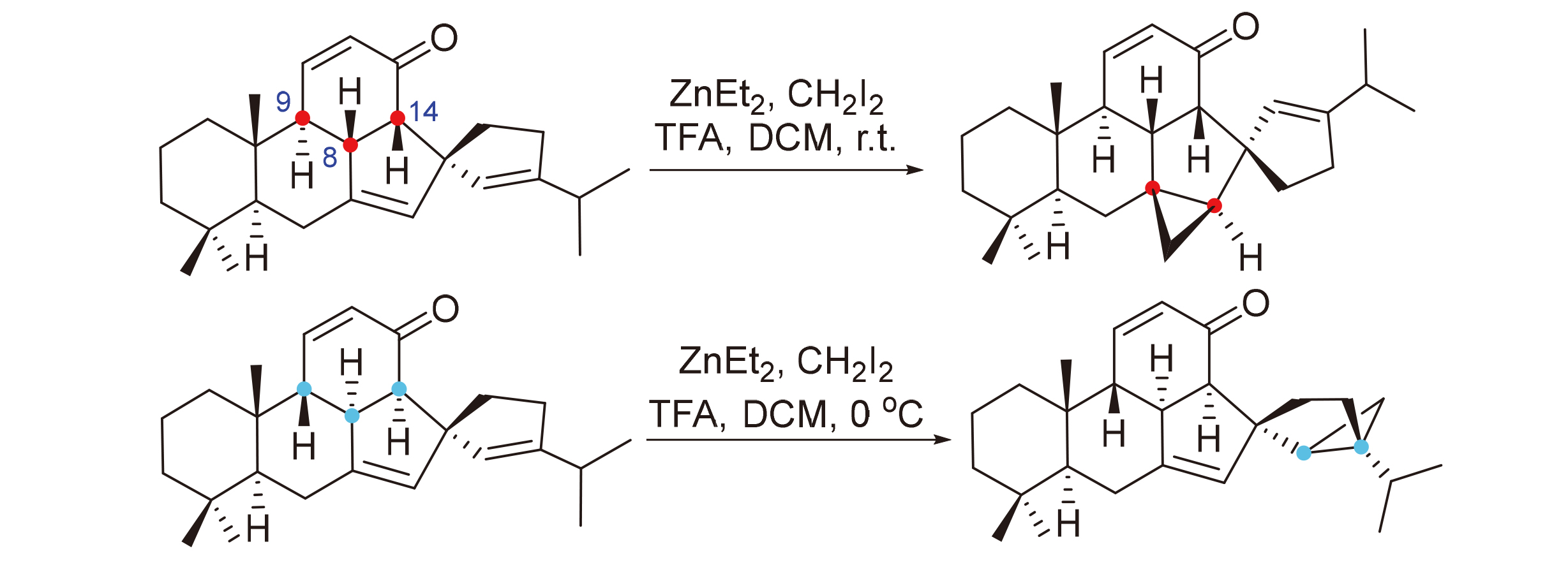
4.3 Cyclopropanation of 8a/b
According to our previously reported procedure,[12] to a stirred solution of Et2Zn (0.411 mL, 1.0 mol/L in hexane, 0.411 mmol, 1.5 equiv.) in dry DCM (2.0 mL) were added successively trifluoroacetic acid (TFA, 31 μL, 0.411 mmol, 1.5 equiv.) and CH2I2 (34 μL, 0.411 mmol, 1.5 equiv.) at 0 ℃ under N2 atmosphere. The solution was stirred for 15 min at the same temperature. Then, the solution of 8a/b (100 mg, 0.274 mmol) in dry DCM (2.0 mL) was added dropwise. The mixture was stirred for 1 h at the same temperature and quenched with water (5 mL). The solution was extracted with DCM (5 mL×3). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification of the residue by column chromatography on silica gel [eluent: V(petro- leum ether)∶V(Et2O)=80∶1] afforded compound 10 (34 mg, 0.090 mmol, 33% yield) as a colorless oil and compound 8a (54 mg, 0.145 mmol, 53% yield) as a colorless oil.
(1'S,3aR,3a1S,4S,5'R,6aS,10aR,10bR)-5'-Isopropyl-7,7,10a-trimethyl-3a,3a1,6,6a,7,8,9,10,10a,10b-decahydro-3H-spiro[acephenanthrylene-4,2'-bicyclo[3.1.0]hexan]-3-one (10): $[\alpha]_{\mathrm{D}}^{20}$+93 (c 0.30, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 7.02 (dd, J=10.2, 2.2 Hz, 1H), 5.90 (dd, J=10.1, 3.3 Hz, 1H), 5.23 (s, 1H), 3.18 (dd, J=11.7, 9.0 Hz, 1H), 3.09 (d, J=8.7 Hz, 1H), 2.55 (d, J=17.3 Hz, 1H), 2.24 (dd, J=17.3, 13.5 Hz, 1H), 2.00 (dt, J=12.1, 2.8 Hz, 1H), 1.76~1.53 (m, 6H), 1.48~1.41 (m, 1H), 1.39~1.34 (m, 1H), 1.32~1.25 (m, 2H), 1.22~1.11 (m, 1H), 1.02 (s, 3H), 1.00~0.89 (m, 14H), 0.89~0.80 (m, 1H), 0.35~0.30 (m, 1H); 13C NMR (75 MHz, CDCl3) δ: 200.5, 149.1, 142.3, 131.0, 130.0, 60.3, 52.1, 52.0, 49.5, 47.6, 41.7, 37.0, 36.0, 34.1, 34.0, 33.8, 32.6, 32.0, 30.4, 27.0, 25.3, 22.3, 22.0, 20.3, 19.9, 19.8, 12.0; HRMS (ESI) calcd for C27H39O [M+H]+ 379.2995, found 379.2987.
(3aS,3a1R,4R,6aS,10aR,10bS)-3'-Isopropyl-7,7,10a-trimethyl-3a,3a1,6,6a,7,8,9,10,10a,10b-decahydro-3H-spiro[acephenanthrylene-4,1'-cyclopentan]-2'-en-3-one(8a): $[\alpha]_{\mathrm{D}}^{20}$-113 (c 0.22, CHCl3); 1H NMR (300 MHz, CDCl3) δ: 6.90 (dd, J=10.1, 2.0 Hz, 1H), 5.93 (dd, J=10.1, 3.0 Hz, 1H), 5.05 (s, 1H), 4.81~4.79 (m, 1H), 2.89~2.80 (m, 2H), 2.46 (dd, J=12.7, 3.5 Hz, 1H), 2.38~2.14 (m, 4H), 2.12~2.00 (m, 2H), 1.87~1.76 (m, 2H), 1.73~1.62 (m, 1H), 1.56~1.41 (m, 2H), 1.26~1.16 (m, 1H), 1.12~1.02 (m, 2H), 0.98 (d, J=6.8 Hz, 6H), 0.95 (s, 3H), 0.91 (s, 3H), 0.89 (s, 3H); 13C NMR (75 MHz, CDCl3) δ: 200.1, 152.8, 147.7, 144.0, 132.6, 128.1, 125.4, 65.1, 58.5, 56.6, 54.6, 48.3, 42.3, 39.2, 37.7, 37.0, 34.0, 33.2, 31.9, 30.0, 25.9, 21.7, 21.5, 21.3, 18.6, 15.9; HRMS (ESI) calcd for C26H37O [M+H]+ 365.2839, found 365.2835.
4.4 Synthesis of (1'S,3aS,3a1R,4S,5'R,6aS,10aS, 10bR)-3,5'-diisopropyl-7,7,10a-trimethyl-3a,3a1,6,6a,7,8,9,10,10a,10b-decahydro-1H-spiro[acephenan-thrylene-4,2'-bicyclo[3.1.0]hexan]-1-one (11)
According to our previously reported procedure,[12] to a stirred solution of 10 (12 mg, 31.7 μmol) in dry THF (2 mL) was added i-PrLi (0.16 mL, 1.0 mol/L in hexane, 0.159 mmol, 5.0 equiv.) dropwise at -78 ℃ under N2 atmosphere. The resulting solution was stirred for 3 h at -78 ℃ and then raised to 0 ℃. The reaction mixture was quenched by water (5 mL) at 0 ℃. The mixture was extracted with EtOAc (5 mL×3). The combined organic layers were washed with brine and dried over anhydrous Na2SO4, filtered, and then concentrated under reduced pressure to afford crude product which was used without purification.
To a stirred solution of the above crude product in DCM (2 mL) was added PCC (13.7 mg, 63.4 μmol, 2.0 equiv.) at 0 ℃. The reaction mixture was stirred for 24 h at room temperature and diluted with water (5 mL). The mixture was extracted with DCM (5 mL×3). The combined organic layers were dried over anhydrous Na2SO4, filtered, and then concentrated under reduced pressure. Purification of the residue by column chromatography on silica gel [eluent: V(petroleum ether)∶V(DCM)=5∶1] afforded compound 11 (9.6 mg, 22.8 μmol, 72% yield over 2 steps) as a white solid. m.p. 120~121 ℃; $[\alpha]_{\mathrm{D}}^{20}$-14 (c 0.10, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 5.78 (s, 1H), 5.21 (s, 1H), 3.21 (dd, J=14.0, 7.5 Hz, 1H), 3.09 (d, J=7.6 Hz, 1H), 2.62 (sept, J=6.8 Hz, 1H), 2.58~2.53 (m, 1H), 2.54~2.48 (m, 1H), 2.19 (dd, J=17.3, 13.7 Hz, 1H), 1.95 (d, J=14.0 Hz, 1H), 1.71~1.61 (m, 2H), 1.65 (dd, J=13.7, 4.3 Hz, 1H), 1.58~1.53 (m, 1H), 1.53~1.49 (m, 1H), 1.46~1.40 (m, 2H), 1.19~1.03 (m, 4H), 1.13 (s, 3H), 1.10 (d, J=7.1 Hz, 3H), 1.09 (d, J=7.1 Hz, 3H), 0.99~0.96 (m, 1H), 0.98 (d, J=6.8 Hz, 3H), 0.91 (d, J=6.8 Hz, 3H), 0.90 (s, 3H), 0.89 (s, 3H), 0.43~0.39 (m, 2H); 13C NMR (126 MHz, CDCl3) δ: 198.6, 168.9, 144.3, 129.8, 125.8, 60.7, 58.9, 49.7, 48.0, 46.5, 42.0, 38.6, 34.6, 34.3, 34.0, 33.4, 33.1, 32.6, 31.7, 31.4, 27.1, 25.1, 23.8, 22.2, 21.1, 20.3, 20.2, 19.9 (×2), 13.5; HRMS (ESI) calcd for C30H45O [M+H]+ 421.3465, found 421.3457.
4.5 Synthesis of (E/Z)-1-(2,4-dinitrophenyl)-2- ((3aS,3a1R,4R,6aS,10aR,10bS)-3'-isopropyl-7,7,10a-trimethyl-3a,3a1,6,6a,7,8,9,10,10a,10b-decahydro-3H-spiro[acephenanthrylene-4,1'-cyclopentan]-2'-en-3-ylidene)hydrazine (13)
To a stirred solution of recovered 8a (18.2 mg, 0.050 mmol) in MeOH (2 mL) were added 2,4-dinitrophenyl- hydrazine 12 (49.5 mg, 0.250 mmol, 5.0 equiv.) and con. H2SO4 (9.8 mg, 0.10 0 mmol, 2.0 equiv.) at 0 ℃. The resulting solution was raised to 50 ℃ and stirred for 18 h at the same temperature. The reaction was quenched by the addition of water (5 mL). The mixture was extracted with EtOAc (5 mL×3), and the combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Purification of the residue by column chromatography on silica gel [V(petroleum ether)∶V(Et2O)=40∶1] afforded compound 13 as a E/Z mixture with ca. 10∶1 ratio (17.4 mg, 0.032 mmol, 64% yield) as a reddish brown solid. 1H NMR for major isomer (500 MHz, CDCl3) δ: 11.40 (s, 1H), 9.12 (d, J=2.5 Hz, 1H), 8.27 (dd, J=9.6, 2.4 Hz, 1H), 7.87 (d, J=9.6 Hz, 1H), 6.67 (dd, J=10.1, 2.2 Hz, 1H), 6.55 (dd, J=10.2, 2.9 Hz, 1H), 5.10 (s, 1H), 4.73 (s, 1H), 3.23 (d, J=8.9 Hz, 1H), 2.63~2.56 (m, 1H), 2.48 (dd, J=12.6, 3.3 Hz, 1H), 2.40~2.15 (m, 5H), 2.07 (t, J=12.4 Hz, 1H), 1.90~1.83 (m, 1H), 1.77~1.67 (m, 1H), 1.66 (d, J=13.7 Hz, 1H), 1.58~1.50 (m, 1H), 1.48~1.42 (m, 1H), 1.25~1.17 (m, 1H), 1.09 (dd, J=12.5, 3.4 Hz, 1H), 1.08~0.98 (m, 1H), 0.96 (s, 3H), 0.92 (s, 3H), 0.90 (s, 3H), 0.84 (d, J=6.8 Hz, 3H), 0.83 (d, J=6.8 Hz, 3H); 13C NMR for major isomer (126 MHz, CDCl3) δ: 153.3, 151.7, 144.9, 143.9, 143.5, 137.7, 130.0, 129.1, 128.2, 126.9, 123.8, 118.8, 116.4, 66.4, 58.5, 55.2, 51.3, 47.5, 42.3, 39.0, 37.9, 37.0, 34.1, 33.3, 32.0, 30.1, 25.9, 21.7, 21.7, 21.4, 18.6, 15.7; HRMS (ESI) calcd for C32H39N4O4 [M-H]- 543.2977, found 543.2980.
4.6 Cyclopropanation of 15a/b
According to our previously reported procedure,[12] to a stirred solution of Et2Zn (0.44 mL, 1.0 mol/L in hexane, 0.440 mmol, 2.5 equiv.) in dry DCM (2.0 mL) were added successively TFA (33 μL, 0.440 mmol, 2.5 equiv.) and CH2I2 (37 μL, 0.440 mmol, 2.5 equiv.) at 0 ℃ under N2 atmosphere. The solution was stirred for 15 min at the same temperature. Then, the solution of 15a/b (64 mg, 0.176 mmol) in dry DCM (2.0 mL) was added dropwise. The mixture was stirred for 1 h at the same temperature and quenched with water (5 mL). The solution was extracted with DCM (5 mL×3). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification of the residue by column chromatography on silica gel [eluent: V(petroleum ether)∶V(Et2O)=80∶1] afforded compound 16 (33 mg, 0.087 mmol, 49% yield) as a colorless oil and compound 15b (23 mg, 0.063 mmol, 36% yield) as a colorless oil.
(1'S,3aS,3a1R,4R,6aS,10aR,10bS)-5'-Isopropyl-7,7,10a-trimethyl-3a,3a1,6,6a,7,8,9,10,10a,10b-decahydro-3H-spiro[acephenanthrylene-4,2'-bicyclo[3.1.0]hexan]-3-one(16): $[\alpha]_{\mathrm{D}}^{20}$-108 (c 0.30, CHCl3); 1H NMR (300 MHz, CDCl3) δ: 6.87 (dd, J=10.1, 1.8 Hz, 1H), 5.96 (dd, J=10.1, 2.9 Hz, 1H), 5.20 (s, 1H), 3.13 (d, J=9.0 Hz, 1H), 2.89 (dd, J=11.4, 9.3 Hz, 1H), 2.44 (dd, J=12.6, 3.4 Hz, 1H), 2.04 (t, J=12.5 Hz, 1H), 1.85~1.80 (m, 1H), 1.76~1.53 (m, 5H), 1.48~1.34 (m, 3H), 1.22~1.17 (m, 1H), 1.06~0.88 (m, 20H), 0.33 (dd, J=7.4, 5.3 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ: 200.4, 148.0, 143.1, 132.7, 129.7, 59.4, 58.5, 54.2, 53.4, 48.6, 42.3, 37.7, 37.0, 34.0, 33.9, 33.3, 32.7, 32.3, 30.4, 27.0, 25.8, 21.3, 20.3, 19.9, 18.6, 15.8, 12.1; HRMS (ESI) calcd for C27H39O [M+H]+379.2995, found 379.2987.
(3aR,3a1S,4S,6aS,10aR,10bR)-3'-Isopropyl-7,7,10a-trimethyl-3a,3a1,6,6a,7,8,9,10,10a,10b-decahydro-3H-spiro[acephenanthrylene-4,1'-cyclopentan]-2'-en-3-one (15b): $[\alpha]_{\mathrm{D}}^{20}$+179 (c 0.10, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 7.02 (dd, J=10.2, 2.2 Hz, 1H), 5.88 (dd, J=10.2, 3.3 Hz, 1H), 5.08 (s, 1H), 4.78~4.75 (m, 1H), 3.14 (dd, J=11.5, 8.7 Hz, 1H), 2.83 (d, J=8.7 Hz, 1H), 2.58~2.50 (m, 1H), 2.34~2.14 (m, 5H), 2.12~2.01 (m, 2H), 1.73~1.65 (m, 3H), 1.58~1.50 (m, 1H), 1.49~1.41 (m, 1H), 1.32~1.25 (m, 1H), 1.19~1.13 (m, 1H), 1.07 (s, 3H), 0.96 (d, J=6.8 Hz, 6H), 0.92 (s, 3H), 0.91 (s, 3H); 13C NMR (75 MHz, CDCl3) δ: 200.1, 153.0, 148.7, 143.5, 130.9, 128.7, 125.0, 66.0, 55.2, 52.2, 49.7, 47.1, 41.7, 39.4, 37.1, 35.6, 34.0, 33.8, 31.8, 30.0, 25.2, 22.2, 22.0, 21.7, 21.5, 19.8; HRMS (ESI) calcd for C26H37O [M+H]+ 365.2839, found 365.2831.
4.7 Synthesis of (1'S,3aR,3a1S,4R,5'S,6aS,10aS, 10bS)-3,5'-diisopropyl-7,7,10a-trimethyl-3a,3a1,6,6a,7,8,9,10,10a,10b-decahydro-1H-spiro[acephenanth-rylene-4,2'-bicyclo[3.1.0]hexan]-1-one (17)
According to our previously reported procedure,[12] to a stirred solution of 16 (42 mg, 0.115 mmol) in dry THF (2 mL) was added i-PrLi (0.58 mL, 1.0 mol/L in hexane, 0.575 mmol, 5.0 equiv.) dropwise at -78 ℃ under N2 atmosphere. The resulting solution was stirred for 3 h at -78 ℃ and then raised slowly to 0 ℃. The reaction mixture was quenched by the addition of water (5 mL) at 0 ℃. The mixture was extracted with EtOAc (10 mL×3). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford the crude product which was used without purification.
To a stirred solution of the above-obtained crude product in DCM (2 mL) was added PCC (49.6 mg, 0.230 mmol, 2.0 equiv.) at 0 ℃. The reaction mixture was stirred for 24 h at room temperature and diluted with water (5 mL). The mixture was extracted with DCM (10 mL×3). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Purification of the residue by column chromatography [eluent: V(petroleum ether)∶V(DCM)=5∶1] afforded compound 17 (32.0 mg, 0.076 mmol, 66% yield over 2 steps) as a white solid. m.p. 134~135 ℃; $[\alpha]_{\mathrm{D}}^{20}$+2.5 (c 0.40, CHCl3); 1H NMR (300 MHz, CDCl3) δ: 5.75 (s, 1H), 5.17 (s, 1H), 3.17 (d, J=7.6 Hz, 1H), 2.99 (dd, J=13.3, 7.7 Hz, 1H), 2.82~2.77 (m, 1H), 2.64 (sept, J=6.8 Hz, 1H), 2.37 (dd, J=12.3, 3.2 Hz, 1H), 1.99 (t, J=12.4 Hz, 1H), 1.96 (d, J=13.3 Hz, 1H), 1.75~1.68 (m, 1H), 1.66~1.57 (m, 2H), 1.46 (sept, J=6.9 Hz, 1H), 1.42~1.36 (m, 2H), 1.32~1.24 (m, 1H), 1.22~1.10 (m, 2H), 1.09 (d, J=6.8 Hz, 3H), 1.08 (d, J=6.8 Hz, 3H), 1.07 (s, 3H), 0.99 (d, J=6.8 Hz, 3H), 0.99~0.96 (m, 1H), 0.94~0.86 (m, 2H), 0.91 (d, J=6.8 Hz, 3H), 0.90 (s, 3H), 0.88 (s, 3H), 0.48~0.41 (m, 2H); 13C NMR (126 MHz, CDCl3) δ: 200.5, 167.6, 144.9, 129.5, 126.4, 60.7, 59.2, 59.1, 50.3, 47.8, 42.4, 38.4, 38.1, 34.2, 33.8, 33.2, 32.6, 32.4, 31.6, 31.2, 27.2, 25.9, 23.9, 21.9, 20.3, 20.2, 20.0, 18.7, 14.5, 13.7; HRMS (ESI) calcd for C30H45O [M+H]+ 421.3465, found 421.3457.
4.8 Synthesis of (E/Z)-1-(2,4-dinitrophenyl)-2- ((3aR,3a1S,4S,6aS,10aR,10bR)-3'-isopropyl-7,7,10a-trimethyl-3a,3a1,6,6a,7,8,9,10,10a,10b-decahydro-3H-spiro[acephenanthrylene-4,1'-cyclopentan]-2'-en-3-ylidene)hydrazine (18)
To a stirred solution of recovered 15b (18.2 mg, 0.050 mmol) in MeOH (2 mL) were added 2,4-dinitrophenyl- hydrazine 12 (49.5 mg, 0.250 mmol, 5.0 equiv.) and con. H2SO4 (9.8 mg, 0.100 mmol, 2.0 equiv.) at 0 ℃. The resulting solution was raised to 50 ℃ and stirred for 18 h at the same temperature. The reaction mixture was quenched by water (5 mL). The mixture was extracted with EtOAc (5 mL×3), and the combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and then concentrated under reduced pressure. Purification of the residue by column chromatography on silica gel [V(petro- leum ether)∶V(Et2O)=40∶1] afforded compound 18 as a ca. 10∶1 E/Z mixture (16.9 mg, 0.031 mmol, 62% yield) as a reddish brown solid. 1H NMR for major isomer (500 MHz, CDCl3) δ: 11.43 (s, 1H), 9.12 (d, J=2.4 Hz, 1H), 8.28 (dd, J=9.6, 2.3 Hz, 1H), 7.89 (d, J=9.6 Hz, 1H), 6.84 (dd, J=10.2, 2.3 Hz, 1H), 6.48 (dd, J=10.2, 3.1 Hz, 1H), 5.13 (s, 1H), 4.70 (s, 1H), 3.17 (d, J=8.6 Hz, 1H), 2.88 (dd, J=11.2, 9.1 Hz, 1H), 2.59~2.53 (m, 1H), 2.40~2.16 (m, 6H), 1.93 (d, J=12.1 Hz, 1H), 1.74~1.66 (m, 3H), 1.64~1.56 (m, 1H), 1.49~1.44 (m, 1H), 1.34~1.26 (m, 1H), 1.22~1.16 (m, 1H), 1.07(s, 3H), 0.93 (s, 3H), 0.92 (s, 3H), 0.82 (d, J=7.3 Hz, 3H), 0.81 (d, J=7.3 Hz, 3H); 13C NMR for major isomer (126 MHz, CDCl3) δ: 153.3, 152.1, 144.9, 144.5, 143.1, 137.7, 130.0, 129.0, 128.9, 126.5, 123.8, 116.9, 116.4, 67.2, 53.1, 50.3, 49.7, 46.4, 41.8, 39.2, 36.9, 36.5, 34.0, 33.9, 32.0, 30.0, 25.6, 22.6, 22.1, 21.7, 21.7, 19.8; HRMS (ESI) calcd for C32H39N4O4 [M-H]- 543.2977, found 543.2978.
4.9 Synthesis of (1S,1a'R,2a'R,2a1'S,5a'R,5b'R, 9a'S,10a'S)-3-isopropyl-5b',9',9'-trimethyl-1',1a',2a',2a1',5a',5b',6',7',8',9',9a',10'-dodecahydro-3'H-spiro-[cyclopentane-1,2'-cyclopropa[f]acephenanthrylen]-2-en-3'-one (19)
To a stirred solution of Et2Zn (0.288 mL, 1.0 mol/L in hexane, 0.288 mmol, 8.0 equiv.) in dry DCM (1 mL) were added successively TFA (21 μL, 0.288 mmol, 8.0 equiv.) and CH2I2 (48 μL, 0.576 mmol, 16.0 equiv.) at 0 ℃ under N2 atmosphere. The resulting solution was stirred for 15 min at the same temperature. Then, the solution of 15b (13.0 mg, 0.036 mmol) in dry DCM (1 mL) was added dropwise. The resulting solution was stirred for 4 h at room temperature and quenched with water (10 ml). The solution was extracted with DCM (5 mL×3), and the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Purification of the residue by column chromatography on silica gel [eluent: V(petroleum ether)∶V(Et2O)=80∶1] afforded compound 19 (11.0 mg, 0.029 mmol, 81% yield) as a white solid. 1H NMR (300 MHz, CDCl3) δ: 6.94 (dd, J=10.3, 1.7 Hz, 1H), 5.88 (dd, J=10.3, 3.1 Hz, 1H), 5.03 (s, 1H), 2.45 (dd, J=11.7, 7.6 Hz, 1H), 2.35~2.30 (m, 1H), 2.28~2.21 (m, 2H), 2.19~2.13 (m, 3H), 2.08~2.00 (m,1H), 1.76~1.68 (m, 2H), 1.61~1.55 (m, 2H), 1.51~1.43 (m, 3H), 1.25~1.21 (m, 2H), 1.17 (s, 3H), 1.11 (dd, J=8.0, 3.8 Hz, 1H), 0.95 (d, J=6.9 Hz, 3H), 0.94 (d, J=6.9 Hz, 3H), 0.91 (s, 3H), 0.86 (s, 3H), 0.68~0.64 (m, 1H), 0.40 (dd, J=8.0, 5.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ: 200.1, 153.6, 150.2, 130.8, 126.2, 60.4, 53.8, 50.2, 47.9, 42.7, 41.0, 36.9, 36.7, 34.7, 33.6, 33.5, 33.0, 31.7, 31.3, 30.0, 26.1, 22.7, 22.3, 21.7, 21.5, 19.8, 16.7; HRMS (APCI) calcd for C27H39O [M+H]+ 379.2995, found 379.2989.
4.10 Synthesis of (1R,1a'S,2a'S,2a1'R,5a'S,5b'R, 9a'S,10a'R)-3-isopropyl-5b',9',9'-trimethyl-1',1a',2a',2a1',5a',5b',6',7',8',9',9a',10'-dodecahydro-3'H-spiro-[cyclopentane-1,2'-cyclopropa[f]acephenanthrylen]-2-en-3'-one (20)
To a stirred solution of Et2Zn (0.328 mL, 1.0 mol/L in hexane, 0.328 mmol, 8.0 equiv.) in dry DCM (1 mL) were added successively TFA (24 μL, 0.328 mmol, 8.0 equiv.) and CH2I2 (55 μL, 0.656 mmol, 16.0 equiv.) at 0 ℃ under N2 atmosphere. The resulting solution was stirred for 15 min at the same temperature. Then, the solution of 8a (15.0 mg, 0.041 mmol) in dry DCM (1 mL) was added dropwise. The resulting solution was stirred for 4 h at room temperature, quenched with water (10 mL) and then extracted with DCM (5 mL×3). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Purification of the residue by column chromatography on silica gel [eluent: V(petroleum ether)∶V(Et2O)=80∶1] afforded compound 20 (12.6 mg, 0.033 mmol, 81% yield) as a white solid. $[\alpha]_{\mathrm{D}}^{20}$-72 (c 0.50, CHCl3); 1H NMR (300 MHz, CDCl3) δ: 7.01 (dd, J=10.2, 2.1 Hz, 1H), 5.86 (dd, J=10.2, 3.0 Hz, 1H), 5.16 (s, 1H), 2.40~2.33 (m, 2H), 2.31~2.19 (m, 2H), 2.15~2.05 (m, 3H), 1.96 (t, J=13.3 Hz, 1H), 1.93~1.87 (m, 1H), 1.77~1.60 (m, 1H), 1.55~1.51 (m, 1H), 1.48~1.42 (m, 1H), 1.30~1.22 (m, 2H), 1.20~1.12 (m, 2H), 1.10~1.03 (m, 2H), 0.97 (d, J=6.8 Hz, 6H), 0.92 (s, 3H), 0.85 (s, 3H), 0.83 (s, 3H), 0.58 (dd, J=5.2, 3.2 Hz, 1H), 0.25 (dd, J=7.9, 5.2 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ: 200.5, 154.1, 150.3, 131.6, 126.3, 59.6, 55.3, 53.8, 51.7, 45.0, 42.4, 38.1, 38.0, 35.7, 35.2, 33.3, 33.1, 30.9, 30.2, 29.8, 28.0, 21.8, 21.7, 21.2, 18.8, 15.4, 12.5; HRMS (APCI) calcd for C27H39O [M+H]+ 379.2995, found 379.2989.
Supporting Information Copies of 1H and 13C NMR, and other 2D NMR spectra for the compounds E-7-D, 8-D, 8a, 10, 11, 13, 15b, and 16~20. The Supporting Information is available free of charge via the Internet at http:// sioc-journal.cn/.
(Cheng, F.)