

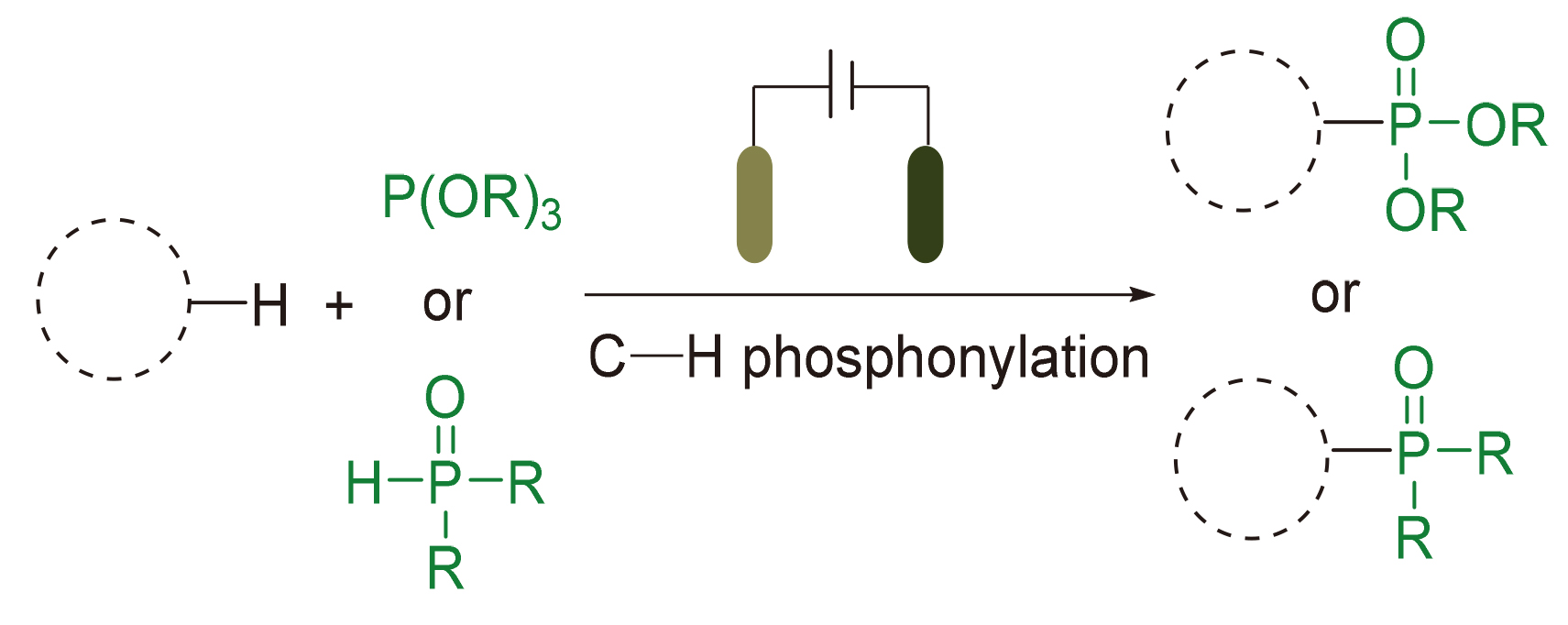
经过国内外多个研究团队的持续探索, (杂)芳烃 C—H键电化学膦酰化反应在底物适用范围、官能团容忍性以及反应的可持续性等方面取得了重要的研究进展. 因此, 该类化合物的高效构建是有机合成化学的研究热点之一. Hirao反应是构建C—P键的经典方法之 一[1], 利用过渡金属催化的(杂)芳基卤代烃与二烷基(或芳基)膦氧化物的偶联, 实现膦酰化的(杂)芳烃的合成. 虽然该方法高效, 但是反应过程中要脱除一分子卤化氢, 降低了反应的原子经济性. 为解决前述方法的局限性, (杂)芳基的直接碳-氢键膦酰化反应在近期的研究工作中获得了显著进展[2]. 和Hirao反应相比, 该策略的原子经济性得到了显著提升. 由于(杂)芳烃与二烷基(或芳基)膦氧化物的偶联是净氧化(net oxidative)反应, 因此需要使用化学氧化剂(比如过硫酸盐等)来促进反应的进行. 虽然可见光促进的(杂)芳基C—H膦酰化策略为上述合成局限性提供了绿色的解决方案[3], 但是, 昂贵光催化剂的使用限制了该类反应的广泛应用.
有机电化学作为一种绿色、高效的合成手段, 在碳-碳以及碳-杂键的构筑中起到了重要的作用[4]. 由于有机电化学采用电子作为清洁的试剂, 可以避免有毒、有害化学氧化还原试剂的使用, 此外, 温和的反应条件有助于提升反应的官能团兼容性[5]. 因此, 有机电化学已成为绿色合成化学的重要工具之一. 鉴于膦酰化的(杂)芳烃的重要用途以及该类化合物合成所存在的问题, 电化学促进的(杂)芳基的C—H膦酰化反应近期取得了重要研究进展. 本综述系统总结了电化学(杂)芳烃C—H膦酰化的重要研究进展, 并对这些方法的应用范围、机理以及目前的局限性进行了评述. 为了便于读者阅读, 我们根据反应机制的不同, 将现有研究方法分为两大类: (1)(杂)芳烃碳-氢键的直接电化学膦酰化反应; (2)过渡金属催化的电化学碳-氢键膦酰化反应. 希望该综述能够为即将投身该领域的青年学者以及研究生提供参考.
1 (杂)芳烃碳-氢键的直接电化学膦酰化反应
芳烃碳-氢键的直接电化学膦酰化反应最早可以追溯到1983年, Nikitin及其合作者[6]利用电化学氧化三烷基亚磷酸酯生成相应的膦自由基, 进而和芳烃发生自由基加成反应, 最终实现芳烃的膦酰化. 但是, 该方法的官能团容忍性一般. 伴随着电化学合成技术的快速发展, (杂)芳烃的直接电化学膦酰化反应在底物适用范围以及官能团容忍性等方面都取得了突破.
2019年, 曾程初课题组[7]利用直接电氧化手段实现了喹喔啉酮类化合物(1)的膦酰化反应(Scheme 1). 该电解反应在单池电解槽中以恒电流电解的方式进行, 石墨和铂网分别作为阳极和阴极材料. 该反应具有较好的官能团容忍性, 例如卤素(3-1), 酯基(3-3)和烯烃(3-4)等官能团都能得到很好的兼容, 并以较好的收率实现 C(sp2)—P键的构筑. 通过控制实验和自由基钟实验等机理研究, 作者认为该反应经历了膦自由基的反应路径. 此外, 循环伏安法实验指出亚磷酸酯的氧化电位要远高于喹喔啉酮. 基于上述机理实验, 作者认为喹喔啉酮的阳离子自由基作为氧化剂, 间接氧化亚磷酸酯生成相应的膦自由基. 首先, 喹喔啉酮在阳极被氧化为相应的阳离子自由基4; 同时, 亚磷酸酯(2)异构化为5. 随后, 阳离子自由基4氧化中间体5生成相应的膦自由基6. 该膦自由基与质子化的喹喔啉酮发生加成反应, 再经后续的氧化与去质子化过程, 最终生成目标产物3. 由于该电解反应使用喹喔啉酮的阳离子自由基(4)氧化亚磷酸酯生成相应的膦自由基, 为膦自由基的电化学生成提供了新的研究思路. 但是, 但该方法具有局限性, 其它的富电子杂环不能参与该膦酰化反应, 主要原因是由于富电子杂环生成的阳离子自由基氧化能力不足以氧化亚磷酸酯.
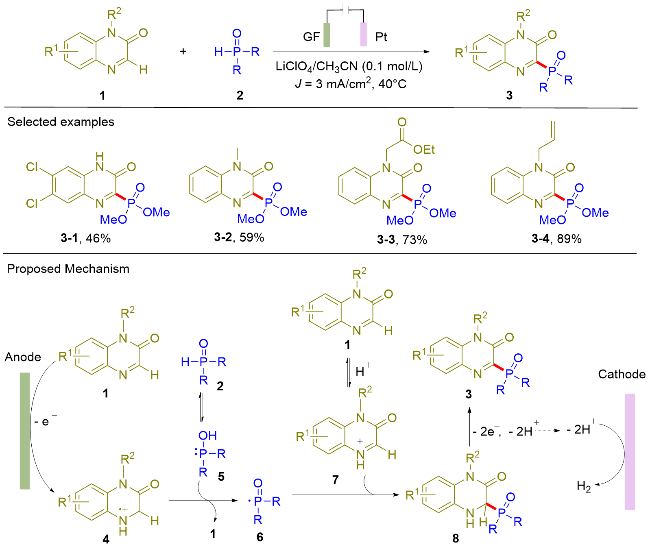
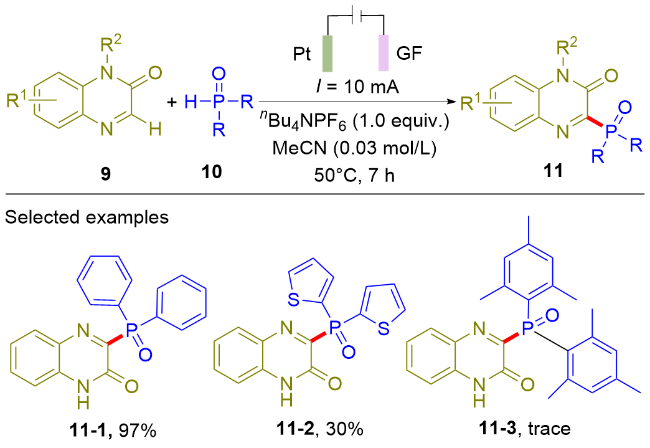
随后, 王利民课题组[8]报道了直接电氧化诱导的喹喔啉酮(9)与二芳基磷氧化物(10)的脱氢偶联反应, 在温和条件下实现了喹喔啉酮C3位的膦酰化反应(Scheme 2). 对于二芳基膦氧化物, 苯基取代的底物能以优异的产率得到目标产物11-1. 然而, 杂环取代的底物只能以30%的收率合成得到目标产物11-2. 作为该类方法的局限性, 大位阻的二芳基膦氧在标准条件下并不能得到目标产物11-3. 根据控制实验和循环伏安实验, 作者认为该反应经历了膦自由基的反应路径. 传统的合成方法需要使用过量的K2S2O8作为氧化剂[9], 相比之下, 本电化学合成方法具有反应过程绿色环保的显著优势. 与Scheme 1所示的电化学合成方法相比, 该策略的优势在于能够以高产率兼容N-未保护的喹喔啉酮类化合物.
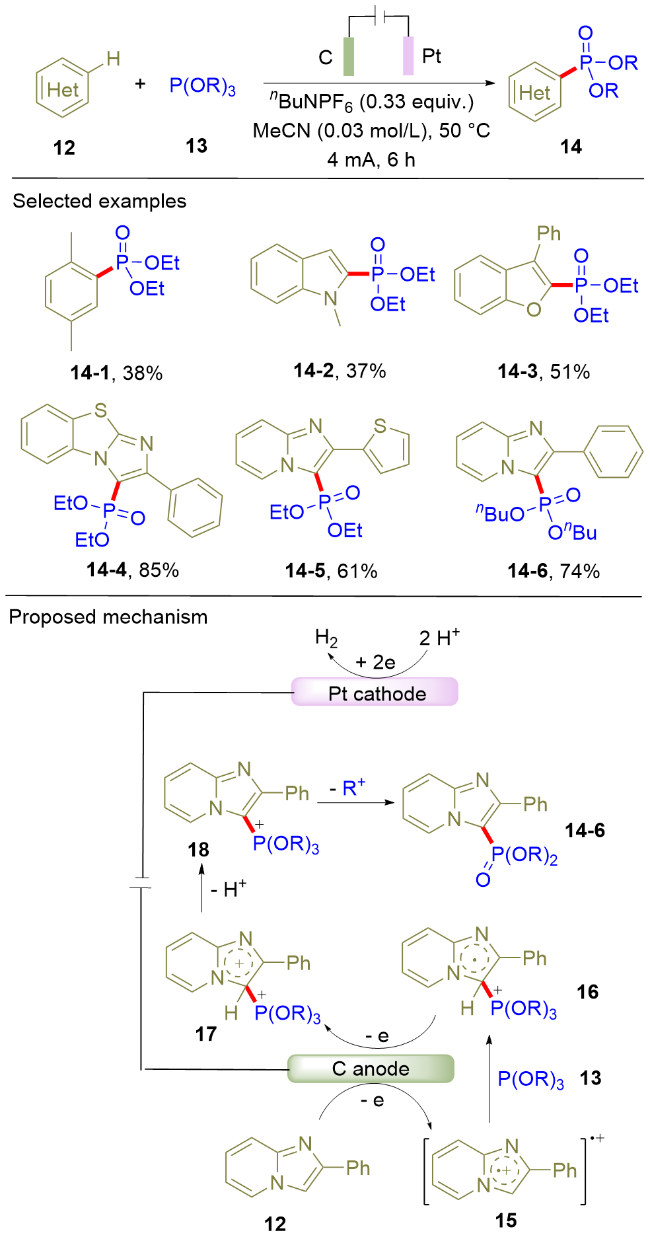
2019年, 雷爱文课题组[10]利用直接电氧化作为绿色合成手段, 实现了(杂)芳烃(12)和亚磷酸三烷基酯(13)的电化学脱氢偶联反应(Scheme 3). 该反应采用石墨和铂片分别作为阳极和阴极材料, 在恒电流电解条件下合成膦酰化的芳香化合物(14). 由于该反应的条件温和, 底物适用范围广. 除了苯环, 一系列杂环化合物比如吲哚、苯并呋喃以及咪唑类化合物都能很好地兼容该反应条件. 根据循环伏安等机理实验, 作者提出了如下的反应路径: 首先, 2-苯基咪唑并[1,2-a]吡啶(12)在阳极发生氧化生成阳离子自由基15; 随后, 该阳离子自由基被亚磷酸三烷基酯捕获生成中间体16; 该中间体发生进一步的阳极氧化和去质子过程生成中间体18; 最后, 中间体18发生脱烃基反应生成膦酰化的杂环化合物14-6. 在上述反应过程中, 阴极发生氢离子的还原, 析出氢气. 由于该反应无需金属催化剂和化学氧化剂的参与, 且反应的唯一副产物为氢气, 因此, 该反应策略具有反应过程绿色、原子经济性高的优势. 值得一提的是, 该方法不但适用于C(sp2)—P键的构筑, 而且还适用于 C(sp3)—P键的构建.
在Scheme 3所示研究策略的基础之上, 2022年, Budnikova课题组[11]实现了吖啶(19)和亚磷酸三烷基酯(20)的电化学脱氢偶联反应(Scheme 4). 该反应以铂片分别作为阳极和阴极材料, 在恒电流条件下电解得到膦酰化产物(21). 值得注意的是, 该反应具有较广的底物适用范围, 除了亚磷酸三烷基酯(21-1, 21-2), (Et2N)2P- OEt和Et2NP(OEt)2也能以中等以上的收率得到目标产物(21-3, 21-4), 但这两种底物在其它的电化学膦酰化反应中无法得到兼容. 通过电子顺磁共振(EPR)实验, 作者提出吖啶和亚磷酸三烷基酯同时在阳极氧化的机理. 此外, 作者还分离得到了膦酰化的二氢吖啶中间体, 并通过单晶衍射确定了该结构, 进一步证实了膦酰化的二氢吖啶到目标产物转化的路径. 由于以往的合成方法需要多步合成才能获取9位膦酰化的吖啶[12], 因此该电化学方法具有显著提升的步骤和原子经济性. 作为该反应的局限性, 由于电解反应需要在H型电解槽中进行, 不利于后续的规模化应用.
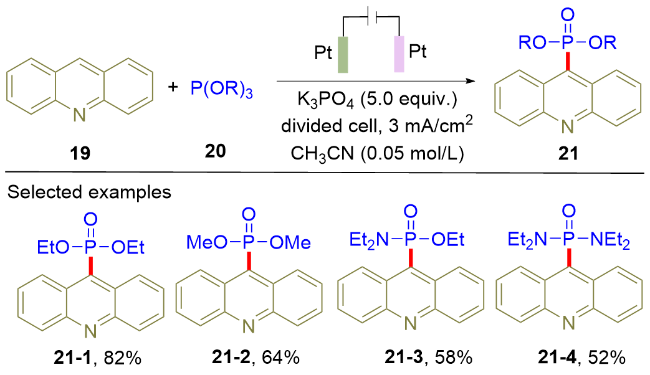
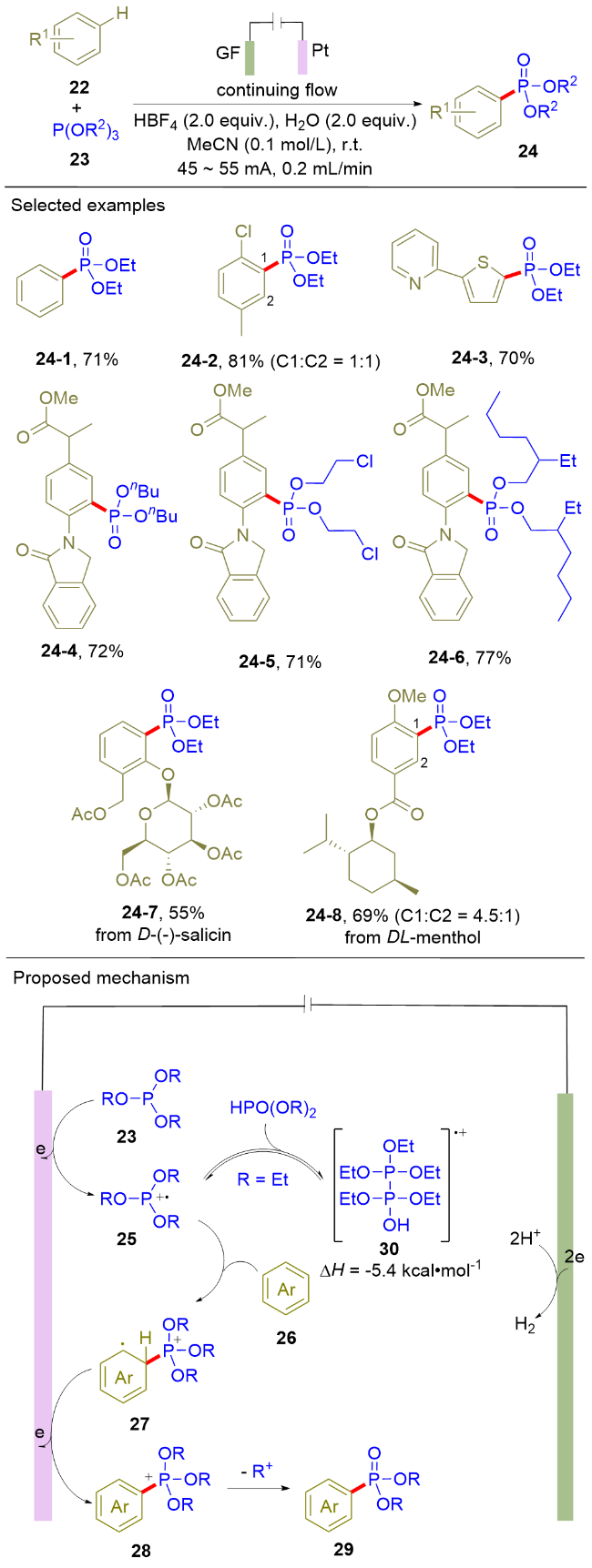
如前所述, (杂)芳烃的直接电化学膦酰化反应已经取得了重要的研究进展, 但是该反应体系存在两点主要局限: 其一, 反应底物富电子与缺电子(杂)芳烃无法同时兼容; 其二, (杂)芳烃常需要过量使用. 为了解决上述问题, 2021年, 徐海超课题组[13]利用连续流动电化学作为技术手段, 实现了富电子以及缺电子(杂)芳烃(22)和亚磷酸三烷基酯(23)的膦酰化反应(Scheme 5). 该流动电解以石墨作为阳极, 铂片作为阴极. 在恒电流的条件下, 一系列(杂)芳烃和亚磷酸三烷基酯顺利地发生脱氢偶联反应, 并以较好的收率获取膦酰化的(杂)芳烃化合物24. 值得注意的是, 该方法对于天然产物衍生的芳烃也能很好地兼容(24-7, 24-8). 循环伏安和核磁标定实验证实了HPO(OR)2在反应中起关键作用; 分子间的动力学同位素效应竞争实验证明芳烃C—H键的裂解不是该反应的决速步骤. 基于以上实验结果, 作者提出了如下的反应路径: 首先, 亚磷酸三烷基酯23在阳极氧化生成相应的含膦阳离子自由基中间体25. 随后, 该中间体被芳烃26捕获生成新的阳离子自由基中间体27, 其在阳极失去电子并伴随去质子化生成膦正离子中间体28. 最后, 中间体28发生脱烷基反应生成目标产物29. 和普通的单池电解槽电解相比, 高达55.0 g目标产物的制备进一步证实该流动电解策略的合成潜力. 与Scheme 3所示的间歇式电解(batch electrolysis)相比, Scheme 5采用的流动电解策略能够以更高产率获得富电子芳烃的膦酰化产物.
2 过渡金属催化的(杂)芳烃电化学碳-氢键膦酰化反应
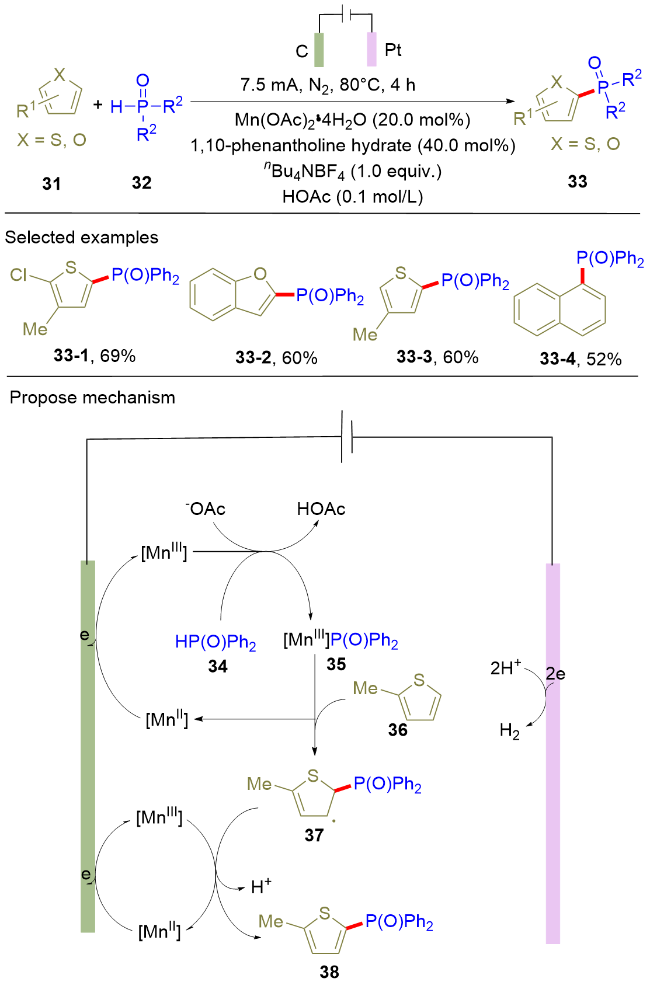
直接电解策略虽然能够高效获取膦酰化的(杂)芳香化合物, 但是直接电解容易造成较高的过电位, 导致某些易氧化的官能团容忍性下降. 为了克服上述局限性, 过渡金属催化的电化学膦酰化反应受到了重点关注. 由于该类电解反应中, 阳极优先氧化电位较低的过渡金属离子, 而不是含膦化合物, 因此, 反应的官能团容忍性得到了进一步提升. 在该种研究策略的指引下, 2021年, 雷爱文课题组[14]实现了锰催化的杂环芳烃(31)与二芳基膦氧化合物(32)的电化学脱氢偶联反应(Scheme 6). 该电解反应在单室电解池中以恒电流电解的方式进行, 以碳棒和铂片分别作为阳极和阴极材料. 在标准条件下, 富电子的芳杂环, 比如噻吩、苯并呋喃和N-甲基-2-乙酰基吡咯等, 均能以中等以上的收率得到膦酰化产物. 除了芳杂环, 萘作为反应底物也可被膦酰化, 并以中等的收率得到目标产物33-4. 作为该反应的局限性, 未取代的吲哚和吡咯在标准条件下并不能参与反应, 可能原因是该类底物会毒化锰催化剂. 同位素效应实验指出芳烃中的C—H键活化不是该反应的决速步骤. 在机理实验的基础上, 作者提出了如下的反应机理: 首先, Mn(II)在阳极氧化为Mn(III), 其与二苯基膦氧(34)结合形成复合物MnIII-P(O)Ph2 (35). 随后, 该复合物将含膦自由基转移至2-甲基噻吩(36), 生成碳自由基中间体37, 并释放Mn(II). 最后, 碳自由基中间体37被Mn(III)氧化, 发生进一步的芳香化生成膦酰化的噻吩化合物38. 1,10-菲咯啉是该反应成功的关键因素之一. 除作为配体稳定锰中间体外, 循环伏安研究表明, 1,10-菲咯啉能够将Mn(II)氧化为Mn(III)的电位从1.9 V降低至1.5 V. 由于该电解反应通过Mn(II)的阳极氧化引发, 该作用有效促进了电解反应的顺利进行.
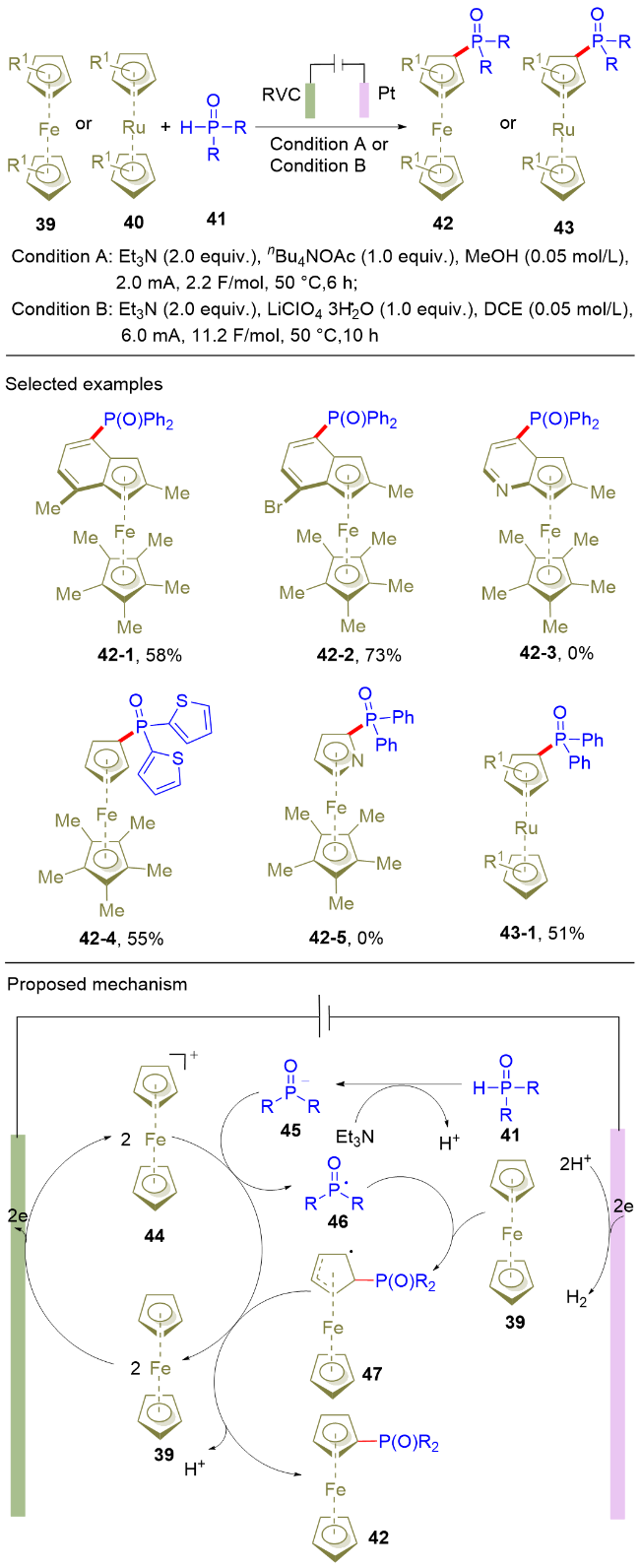
基于茂金属的含膦化合物, 在有机合成中常被用作配体或者催化剂, 因此该类化合物的合成具有重要的研究意义. 在已发展的合成方法中, 茂金属C—H键的直接官能化是合成上述化合物的最有效手段之一. 相较于对称的双环戊基茂金属的C—H官能化, 非对称的茚基茂金属的C—H官能化至今仍是该领域极具挑战性的难题之一, 主要原因在于难以实现优异的位置选择性控制. 针对上述问题, 陈庆安课题组[15]创造性地实现了茂金属化合物的电化学区域选择性膦酰化反应(Scheme 7). 该电解反应分别使用RVC和铂片作为阳极和阴极, 反应在单池电解槽中以恒电流电解的方式进行. 对于非对称的茚基茂金属, C—H膦酰化具有优异的区域选择性. 除了二茂铁类化合物, 二茂钌类化合物也能很好地进行C—H膦酰化反应, 并以51%的收率得到目标产物(43-1). 除了二苯基膦氧化合物, 二噻吩膦氧化物也能以中等收率制备相应的膦酰化产物. 遗憾的是, 当茂金属中的碳原子被氮原子取代, 无法得到目标产物(42-5). 机理研究表明: 茂金属不仅作为反应底物, 同时还作为电催化剂. 首先, 二茂铁(39)在阳极氧化变为Fe(III)化合物44, 其作为间接氧化剂氧化二芳基膦氧负离子45, 生成相应的膦氧自由基中间体46. 随后, 膦氧自由基与二茂铁39发生自由基加成反应, 生成碳自由基中间体47. 最后, 中间体47发生氧化反应生成膦酰化目标产物42. 在反应过程中, 阴极发生析氢反应, 因此该反应具有较好的原子经济性. 该电化学方法为膦酰化的茂金属制备提供了全新且便捷的途径, 同时也为非对称茂金属的C—H选择性官能团化提供重要的参考价值.
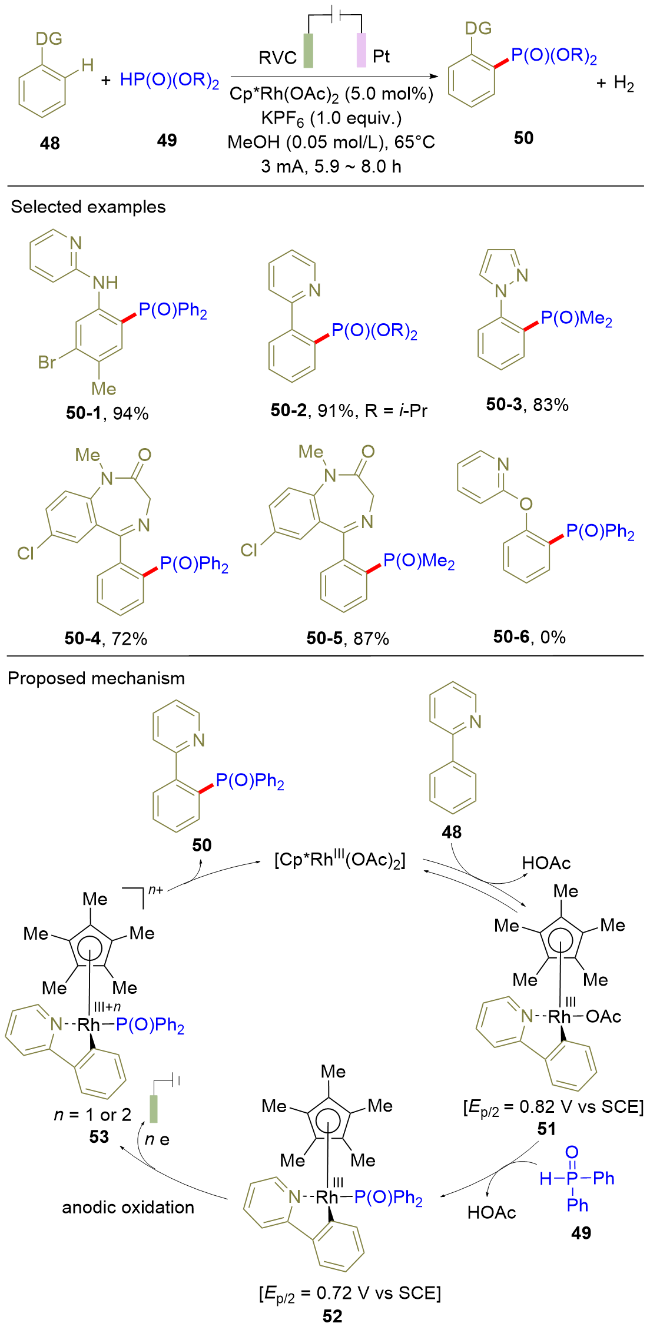
过渡金属催化的导向C—H官能团化是实现区域选择性官能化的重要策略之一. 2019年, 徐海超课题组[16]借助该策略, 利用电化学促进的Rh(III)催化, 实现了芳烃的导向C—H膦酰化反应(Scheme 8). 该反应在单室电解池中以恒电流模式进行电解, 以RVC和铂分别作为阳极和阴极材料. 在标准条件下, 2-氨基吡啶、吡啶以及唑都可以作为导向基团, 实现芳烃邻位的C—H膦酰化反应. 但是, 当以2-羟基吡啶作为导向基团时, 反应不能发生. 通过机理研究, 作者提出了如下的反应机理: 首先, Rh(III)活化2-苯基吡啶48的C—H键生成Rh(III)中间体51, 随后该中间体51与二芳基膦氧进行配体交换生成Rh(III)中间体52. 接下来, 该中间体52发生进一步的电氧化反应生成更高价态的Rh中间体53. 最后, 该中间体53发生还原消除反应生成目标产物50, 并伴随Rh(III)的循环. 该策略实现了过渡金属催化的导向 C—H膦酰化反应, 为常规方法难以合成的三芳基氧化膦的制备提供了新的研究思路.
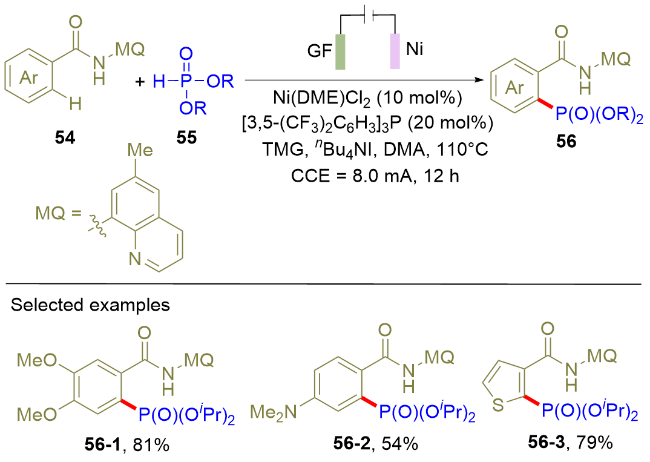
在上述研究工作的启发下, 2021年, Ackermann课题组[17]报道了电化学促进的镍盐催化的导向C—H膦酰化反应(Scheme 9). 该反应在单室电解池中, 以恒电流模式电解, 以石墨和泡沫镍分别作为阳极和阴极材料. 以8-氨基喹啉作为导向基团, 多种取代基类型的芳烃都可以发生C—H键的膦酰化反应, 并以中等以上的收率制备目标化合物. 除了芳烃, 杂环芳烃比如噻吩衍生物也可以发生区域选择性的C—H膦酰化反应. 同位素效应实验结果显示, 芳烃的C—H键活化是该反应的决速步骤. 作者通过高分辨质谱检测到了Ni(IV)和Ni(II)中间体. 在上述实验的基础上, 结合循环伏安实验与理论计算结果, 作者提出该偶联反应经历了Ni(III/IV/II)的催化循环. 与Scheme 5的以Rh作为催化剂的方法相比, 该策略使用廉价的Ni盐作为催化剂, 具有成本经济的优势. 四丁基碘化铵是该反应不可或缺的添加剂, 但是该添加剂的作用目前尚不明确, 因此, 更加深入的机理研究是十分必要的.
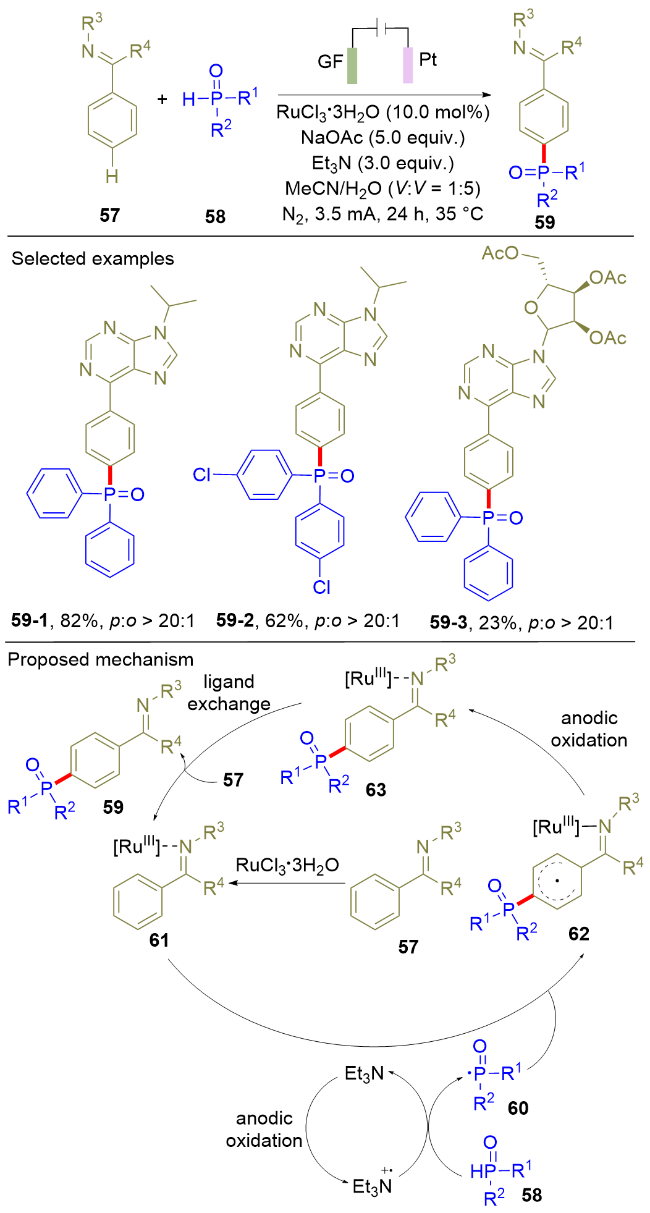
导向的过渡金属电催化可以很好地实现芳烃的邻位C—H膦酰化反应, 如Schemes 8和9所示. 但是, 芳烃的对位选择性C—H膦酰化反应至今仍是该领域极具挑战性的难题. 为了解决该难题, 2025年, Ackermann课题组[18]利用电化学氧化作为手段, 实现了Ru催化的芳烃对位选择性C—H膦酰化反应(Scheme 10). 该电化学方法对芳烃类底物展现出较广的适用性, 其对/邻位选择性可达20∶1以上. 然而, 该方法仍存在一定局限性, 具体表现为二烷基膦氧化合物不能参与反应. 自旋捕获实验和自由基钟实验结果显示, 该反应过程中有膦中心自由基产生. 根据机理实验结果, 作者提出了如下的反应机理: 首先, 三乙胺在阳极氧化为相应的阳离子自由基, 其作为氧化剂促进二芳基膦氧58到相应膦自由基60的转化. 随后, 膦自由基与Ru(III)复合物发生加成反应生成碳中心自由基62, 由于Ru(III)的配位作用, 自由基加成选择性地在对位发生. 最后, 碳自由基62发生进一步的阳极氧化反应, 并伴随配体交换, 生成目标化合物59. 该电催化策略为芳烃的对位选择性官能化提供了重要的借鉴意义.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
经过国内外多个研究团队的持续探索, (杂)芳烃 C—H键电化学膦酰化反应已取得一系列重要研究进展. 本文系统综述了该领域的最新研究成果, 基于反应所采用的电解模式差异, 将相关反应体系划分为两大类. 与传统合成方法相比, 电化学策略在原子经济性和官能团兼容性方面展现出显著优势. 特别值得关注的是, 相较于直接电化学膦酰化反应, 过渡金属催化的电化学策略在反应选择性方面表现更为突出, 尤其是导向基团辅助的C—H膦酰化反应.
尽管该研究领域已取得重要进展, 但仍面临诸多挑战. 基于现有研究, 我们认为未来发展方向重点关注以下方面: 首先, 当前电解体系通常依赖大浓度的电解质时, 流动电解技术的应用有望显著降低其用量. 其次, 过渡金属催化体系中普遍存在催化剂用量偏高的问题, 主要归因于金属在阴极的沉积失活, 而引入交流电解策略可有效降低催化剂负载量. 最后, 在导向C—H膦酰化反应领域, 现有研究主要集中于邻位选择性, 而对位选择性转化至今仍是亟待突破的关键科学挑战, 具备重要的研究价值.
(Cheng, F.)