

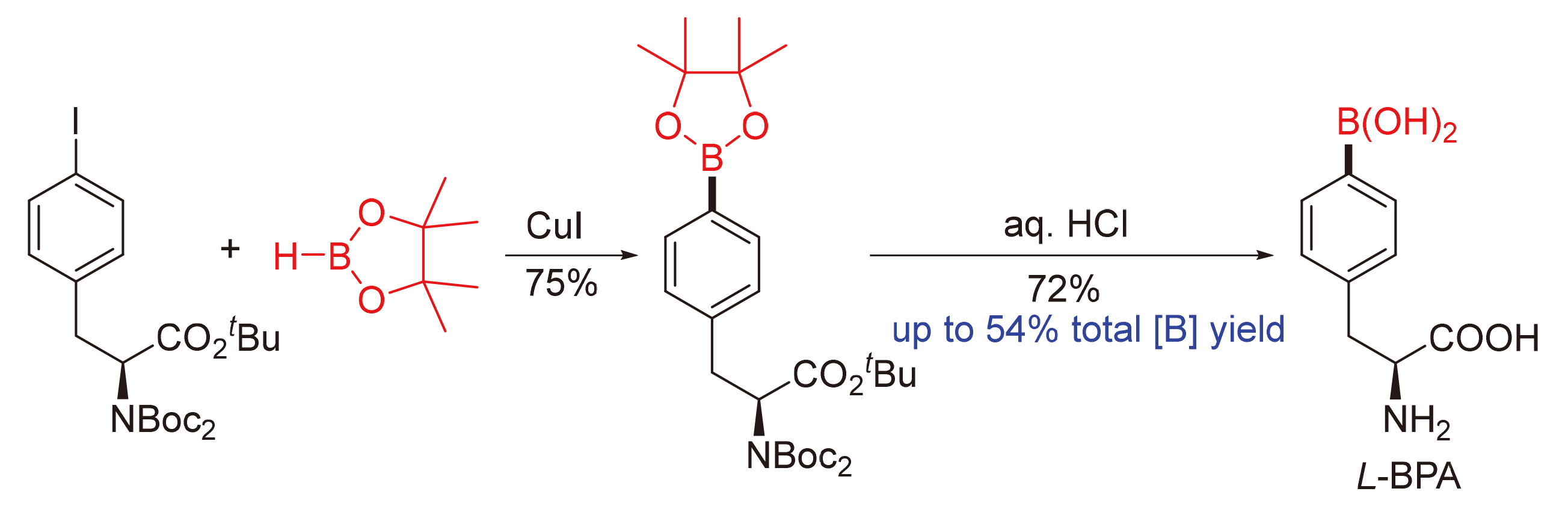
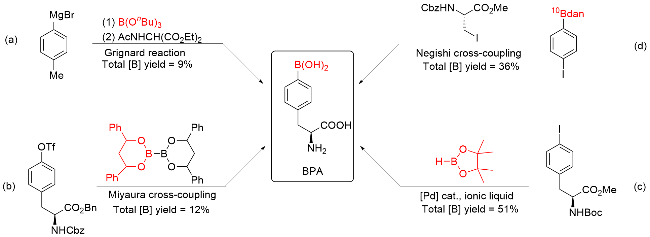
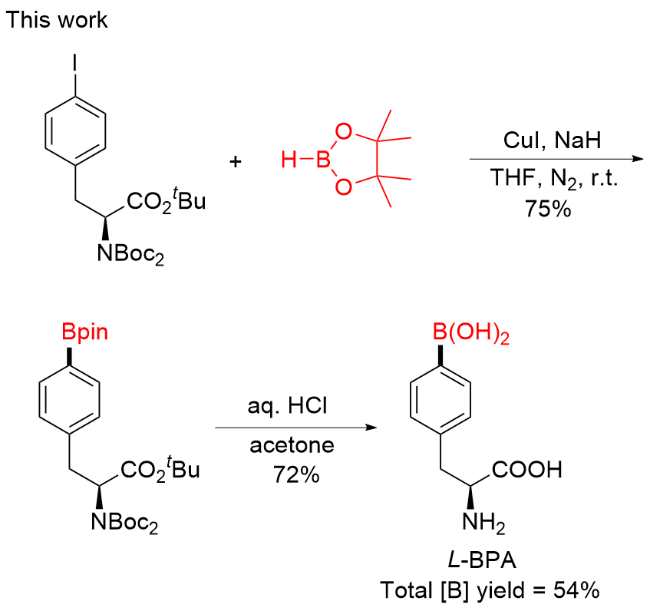
1 结果与讨论
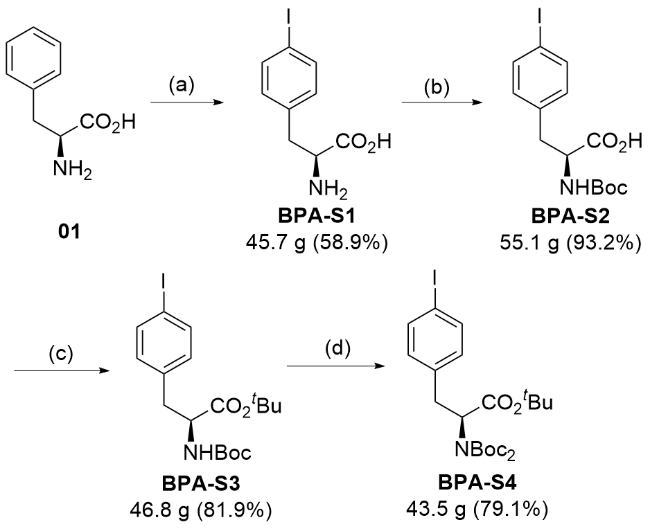
图4 中间体BPA-S3和BPA-S4的合成路线Figure 4 Synthetic route of intermediates BPA-S3 and BPA-S4 Reaction reagents and conditions: (a) I2, NaIO3, AcOH/H2SO4, 70 ℃, 20 h, then NaIO4, 70 ℃, 4 h, 59%; (b) NaHCO3, Boc2O, THF/H2O, r.t., 4 h, 93%; (c) Boc2O, DMAP, tBuOH, r.t., 5 h, 82%; (d) Boc2O, DMAP, THF, 50 ℃, 4 h, 79%. |
表1 反应条件优化aTable 1 Optimization of the reaction conditions 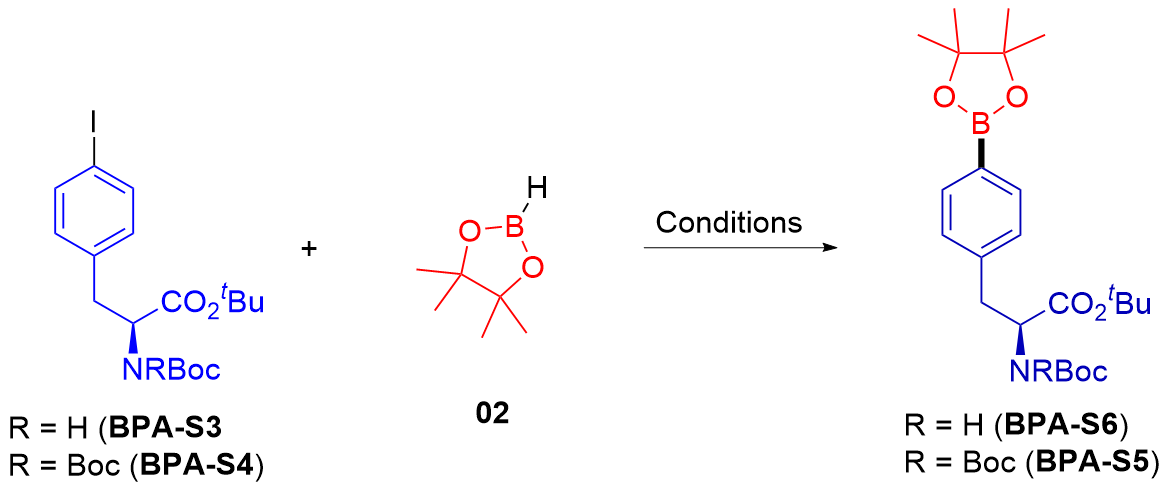 |
| Entry | Catalyst | Additive | Solvent | Temperature/oC | Yield/% |
|---|---|---|---|---|---|
| 1 | Pd(dppf)Cl2 | Et3N | Dioxane | 100 | N.R. |
| 2 | Ni(dppf)Cl2 | Et3N | Toluene | 100 | N.R. |
| 3 | Pd(OAc)2/DPEphosb | Et3N | Dioxane | 100 | N.R. |
| 4 | CrCl3/dtbpyb | Mg | THF | 90 | N.R |
| 5 | Cp2TiCl2 | MeOLi | Neat | 100 | N.R |
| 6 | Pd(dppf)Cl2 | TMEDA | Dioxane | 100 | N.R |
| 7 | Pd(dppf)Cl2 | tBuOK | Dioxane | 100 | N.R |
| 8 | Pd(dppf)Cl2 | Et3N | DMSO | 100 | N.R. |
| 9 | CuI | NaH | THF | r.t. | 64 |
| 10 | CuIc | NaH | THF | r.t. | 80 |
| 11 | CuIc | NaH | THF | 40 | 75 |
| 12 | CuIc | NaH | THF | 55 | 73 |
| 13 | CuIc | NaH | THF | Reflux | 74 |
| 14 | CuIc | tBuOK | THF | r.t. | 23 |
| 15 | CuIc | Cs2CO3 | THF | r.t. | 19 |
| 16 | CuIc, d | NaH | THF | r.t. | 50 |
| 17 | CuI+Fe(acac)3e | NaH | THF | -10 | N.R. |
| 18f | Pd(dppf)Cl2 | Et3N | Dioxane | 100 | N.R. |
| 19f | Pd(OAc)2/DPEphosb | Et3N | Dioxane | 100 | N.R. |
| 20f | CuIc | NaH | THF | r.t. | N.R. |
a Reaction conditions: BPA-S4 (5.0 mmol), 02 (6.0 mmol), catalysis (10 mol%), additive (7.5 mmol), solvent (20 mL), N2, 3 h. b The amount of the ligand is 2.0 equiv. of the catalyst. c Activated CuI. d CuI (5 mol%). e Fe(acac)3 (10 mol%). f BPA-S3 (5.0 mmol), 02 (6.0 mmol), additive (7.5 mmol), catalysis (10 mol%), solvent (20 mL), 3 h, N2. |
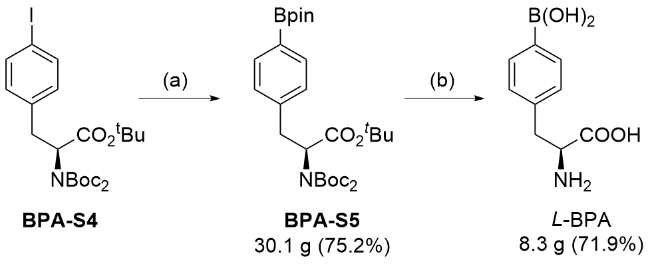
图5 L-BPA的合成路线Figure 5 Synthetic route of L-BPA Reaction reagents and conditions: (a) HBpin, activated CuI, NaH, THF, r.t., 8 h, N2, 75%; (b) aq. HCl (4 mol/L), acetone, reflux, 5 h, 72% |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
表2 L-BPA (10B)路线成本核算Table 2 L-BPA (10B) route cost accounting |
| Entry | Synthetic route of L-BPA (¹⁰B) | Synthetic step | Boron utilization rate/% | Total material cost/ (104 yuan•kg-1) |
|---|---|---|---|---|
| 1 | Synthetic method reported by the Snyder group[10] | 7 | 9 | 20.4 |
| 2 | Synthetic method reported by the Yamamoto group [11] | 6 | 12 | 17.3 |
| 3 | Synthetic method reported by the Zaidlewicz group[12] | 6 | 51 | 7.5 |
| 4 | Synthetic method reported by the Wakamiya group [13] | 7 | 36 | 8.1 |
| 5 | Synthetic method in this work | 6 | 54 | 6.3 |
2 结论
3 实验部分
3.1 仪器与试剂
3.2 实验方法
3.2.1 化合物BPA-S1的合成
3.2.2 化合物BPA-S2的合成
3.2.3 化合物BPA-S3的合成
3.2.4 化合物BPA-S4的合成
3.2.5 化合物BPA-S5的合成
3.2.5 化合物L-BPA的合成
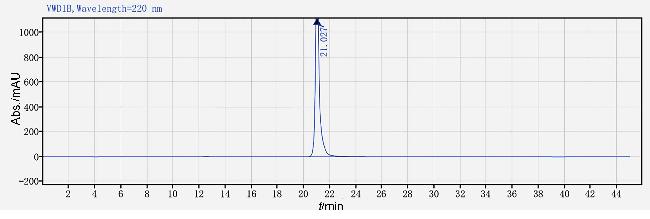
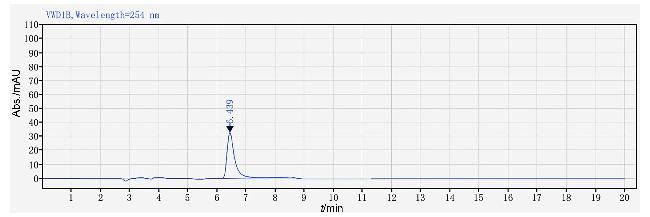
3.2.6 化合物L-BPA的液相分析
表3 流动相A和流动相B的梯度洗脱条件Table 3 Mobile phase composition of gradient elution |
| t/min | Mobile phase A/% | Mobile phase B/% |
|---|---|---|
| 0 | 10 | 90 |
| 3 | 10 | 90 |
| 28 | 30 | 70 |
| 35 | 30 | 70 |
| 35.1 | 10 | 90 |
| 45 | 10 | 90 |