


The cyclization reaction of 1,n-enynes is highly valued by chemists for its atom economy, high efficiency, and excellent functional group compatibility.[1] It is a key synthetic method for rapidly obtaining complex cyclic compounds. Among these reactions, N-(2-ethynylphenyl)acryl-amides stand out due to their multiple reaction sites, making them essential precursors for various functional molecules in synthetic chemistry.[2] The cyclization of N-(2-ethynyl-phenyl)acrylamides facilitates the simultaneous formation of multiple new chemical bonds in a single step and allows the selective introduction of various external functional groups. This methodology is essential for the construction of intricate cyclic compounds, including those with heteroatoms like nitrogen, sulfur, and oxygen. It offers an efficient strategy for the synthesis of polycyclic compounds, significantly advancing the fields of organic and medicinal chemistry. This manuscript provides a comprehensive overview of recent advances in tandem cyclization reactions involving N-(2-ethynylphenyl)acrylamides. This review is organized based on the structure of the resulting products, aiming to serve as a valuable reference for researchers in this field.
In general, the synthesis of N-(2-ethynylphenyl)acryl-amides follows a general procedure as illustrated in Scheme 1. Starting from readily available substituted ortho-amino-iodobenzenes, a Sonogashira coupling reaction with alkynes is performed under the catalysis of palladium and copper to yield compound 1A.[3] Compound 1A then undergoes N-acylation with various acyl chlorides to form compound 1B. Finally, compound 1B is treated with NaH to remove the hydrogen atom and reacts with alkyl halides to produce N-(2-ethynylphenyl)acrylamides.[4]
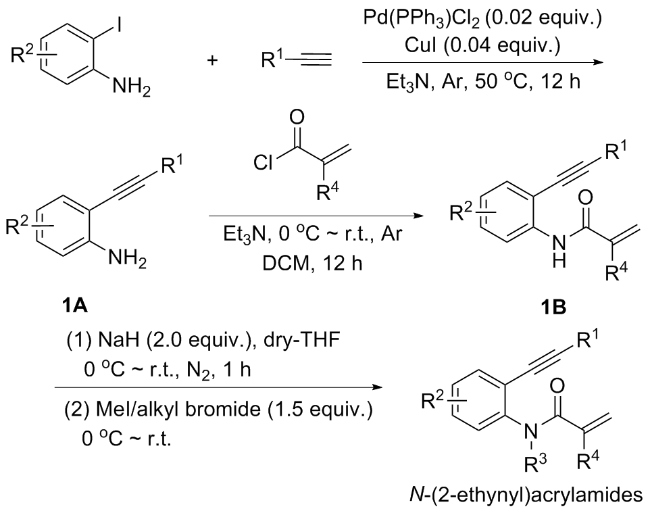
1 Formation of a fused six/N-three-mem- bered ring
In 2017, Han’s group[5] introduced an innovative approach for the synthesis of trifluoromethylated 1'H-spiro- [azirine-2,4'-quinolin]-2'(3'H)-ones, achieved through a CF3-radical-mediated tandem reaction of N-(2-ethynyl-phenyl)acrylamides. This method employed 1-trifluorome-thyl-1,2-benziodoxole as the trifluoromethylating agent and TMSN3 as the aminating agent. Despite the anticipation, the targeted reaction remained unsuccessful with the unprotected amide, potentially attributable to the pivotal role of N-protecting groups in fostering a predominant population of the reactive rotamer configuration, wherein the radical center assumes a proximal stance to the triple bond, thus enhancing intramolecular annulation. Moreover, the current strategy encountered limitations when applied to phenyl- substituted substrates and those devoid of allylic substitution. Regrettably, alkyl-substituted alkynes likewise exhibited a lack of responsiveness within the confines of this methodology. The ensuing transformation of intermediate 2B can unfold via two pathways. The first pathway involves the cyclization of 2B to generate radical 2C, followed by azidation catalyzed by Cu(II) to yield intermediate 2E. Subsequently, denitrogenative cyclization transforms 2E into the desired compound. Alternatively, in the second pathway, Cu(II) intercepts radical 2B prior to cyclization, directly resulting in the formation of intermediate 2E (Scheme 2).
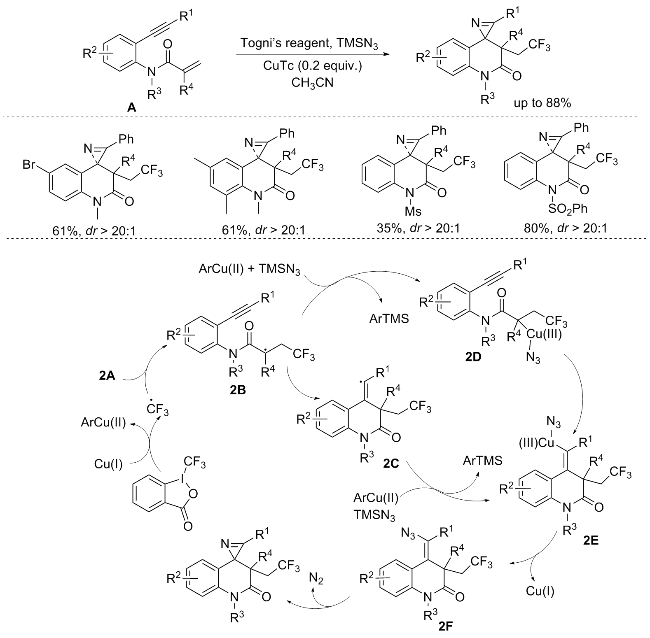
In the same year, a similar reaction was reported by Shi’s group,[4b] utilizing the Togni’s reagent and TMSN3 to develop a copper-catalyzed sequential trifluoromethylation and rearrangement of N-(2-ethynylphenyl)acrylamides. This strategy offers a facile route to structurally diverse and highly valuable trifluoromethylated 1'H-spiro[azirine-2,4'-quinolin]-2'(3'H)-ones, thereby significantly enriching the synthetic arsenal within this domain. Notably, this reaction demonstrates remarkable diastereoselectivity, along with good isolated yields. However, it is noteworthy that aliphatic and pyridyl alkynes failed to yield the desired products, highlighting potential limitations in substrate compatibility. Nevertheless, the obtained products, which are amenable to scalable synthesis, can undergo further transformation via reduction with NaBH4, affording a diverse array of synthetically significant furoindolines featuring three contiguous quaternary carbon centers (Scheme 3).
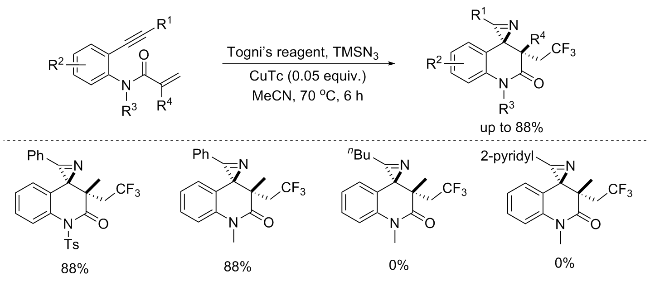
The aforementioned reactions utilized N-(2-ethynyl-phenyl)acrylamides as substrates, with Togni’s reagent and TMSN3 serving as reactants to respectively give the trifluoromethyl group and nitrogen atoms. Under various copper-catalyzed conditions, a radical cascade cyclization occurs, yielding structurally diverse trifluoromethylated 1'H-spiro[azirine-2,4'-quinolin]-2'(3'H)-ones. These reactions provided effective method for synthesizing potentially bioactive trifluoromethylated quinolone compounds. The presence of nitrogen-containing three-membered rings in the products also opens up possibilities for further derivatization.
2 Formation of a fused six/five-membered ring
2.1 Formation of six/N-five-membered ring
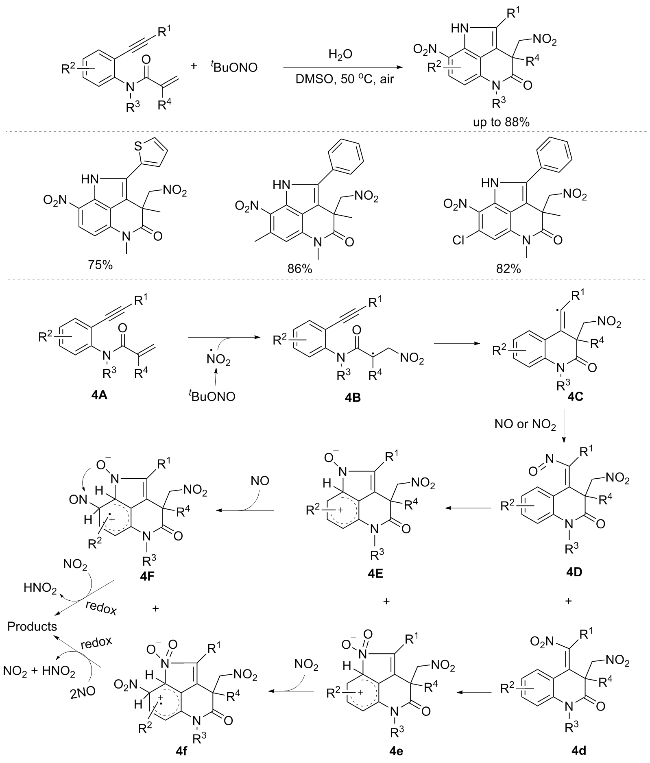
Pyrroloquinolinones are vital components of various natural compounds and pharmaceutical scaffolds, exhibit-ing significant biological and medicinal properties. They are widely utilized as valuable functional intermediates. In 2014, the Li’s group[6] realized one-pot synthesis of the pyrrolo[2-de]quinolinone core structure, through a sequential nitration/cyclization cascade reaction, involving 1,7-enynes with t-BuONO and H2O. The substituents at the ortho-, meta-, or para-positions do not significantly influence the reaction. Regrettably, this reaction is not applicable to aliphatic alkynes. To elucidate the reaction mechanism, they introduced three radical scavengers of 2,2,6,6-tetrame-
thylpiperidoxyl (TEMPO), hydroquinone, and butylhydroxytoluene (BHT), each at a concentration of 4 equiv., into the reaction mixture. This strategic manipulation resulted in a notable inhibition of the reaction, indicative of the involvement of a radical intermediate in the reaction pathway. This hypothesis was reinforced by complementary evidence garnered from an intermolecular kinetic isotope effect (KIE) experiment, further validating the role of the radical species as a crucial intermediate in the transformation. An 18O-labeling experiment with H218O was conducted, indicating that nitration at the 4-position of the N-phenyl moiety is crucial for the described reaction. Furthermore, it was demonstrated that H2O is not the sole source of the oxygen atoms in the nitro groups. The proposed reaction mechanism was illustrated in Scheme 4. In the initial stage of the reaction, the in situ generation of NO2 from t-BuONO prompts its addition to the carbon-carbon double bond of the 1,7-enyne 4A, resulting in the formation of an alkyl radical intermediate 4B. This intermediate 4B undergoes cyclization, yielding intermediate 4C. Subsequently, the reaction of intermediate 4C with either NO or NO2, as evidenced by HRMS analysis, furnishes the corresponding intermediates 4D and 4d. The electrophilic addition of the N=O group to the phenyl ring within intermediates 4D and 4d leads to the formation of cationic intermediates 4E and 4e. Notably, upon subsequent treatment of these cationic intermediates with NO or NO2, a selective transformation occurs, affording cationic radical intermediates 4F and 4f, respectively. Ultimately, the redox reaction involving the cationic radical intermediates 4F and 4f culminates in the production of the desired product. (Scheme 4).[6]
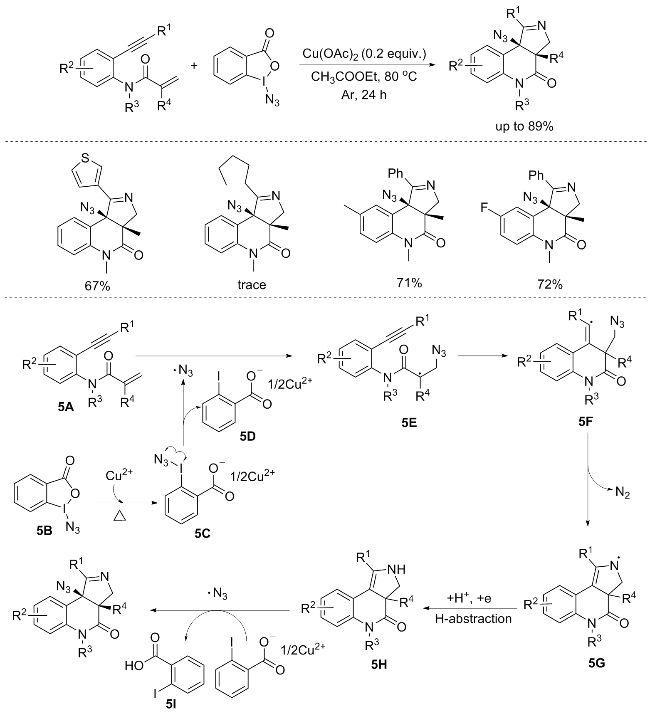
A selective copper-catalyzed radical [2+2+1] annulation process was established by Li’s group, enabling the synthesis of fused pyrroline compounds from benzene-linked 1,n-enynes and azidobenziodoxolone. This method offers an efficient and pragmatic route for the strategic incorporation of a nitrogen atom derived from azidobenziodoxolone into a five-membered cyclic framework. However, it is noteworthy that aliphatic alkynes displayed a lack of reactivity towards this specific reaction. Notably, the reaction between the 1,7-enyne and azidobenziodoxolone was comprehensively quenched when a stoichiometric quantity of radical scavengers, specifically TEMPO, hydroquinone, and BHT, was introduced. This observation underscores the pivotal role of a radical pathway in this transformation, as evidenced by the complete suppression of reactivity under these conditions.
The reaction mechanism was shown in Scheme 5. Initially, the generation of an azide radical from azido-benzio-doxolone 5B, facilitated by the activation of Cu2+ species under thermal conditions, initiates the reaction sequence. This process leads to the formation of N3-substituted 2- iodobenzoate intermediate 5C, which subsequently undergoes decomposition, yielding a stable azide radical and copper(II) salt 5D. Subsequently, the addition of this azide radical to the alkene moiety of enyne 5A results in the formation of alkyl radical intermediate 5E. This intermediate 5E readily undergoes intramolecular cyclization with the adjacent alkyne moiety, generating vinyl radical intermediate 5F. Subsequently, an intramolecular addition of the vinyl radical to an azido group within 5F occurs, followed by the elimination of N2, yielding an aminyl radical intermediate 5G. This aminyl radical 5G then undergoes transformation into intermediate 5H, either through a hydrogen abstraction mechanism or a reduction-protonation sequence. Finally, the addition of another azide radical to the alkene moiety of intermediate 5H, followed by deprotonation catalyzed by 2-iodobenzoate 5D, culminates in the formation of the product, accompanied by the regeneration of 2-iodobenzoic acid 5I (Scheme 5).[7]
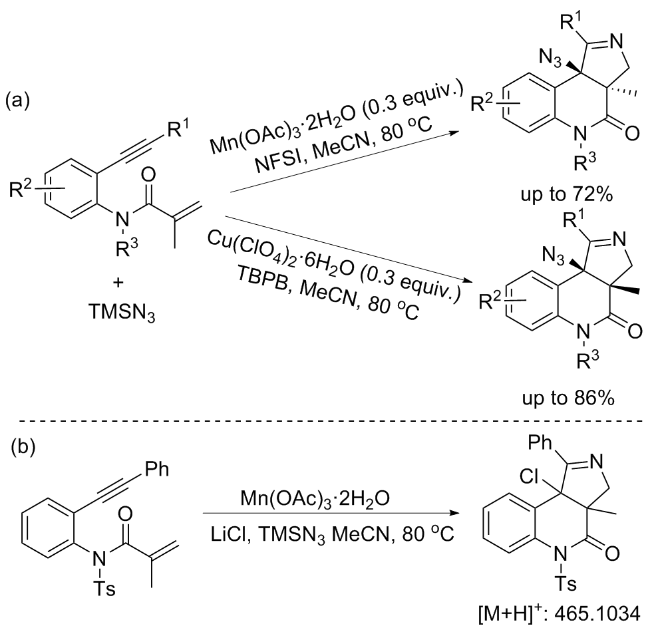
In the following year, the cyclization reaction between N-(2-ethynylphenyl)acrylamides and TMSN3 was also achieved by Wan’s group.[8] Notably, the diastereoselecti- vity was successfully manipulated by adjusting the transition-metal catalysts and their corresponding ligands. Specifically, the Mn(III)-mediated radical cyclization/azidation cascade reaction of N-(2-ethynylphenyl)acrylamides yie- lded trans-fused pyrrolo[3,4-c]quinolinones, whereas the Cu(II)/bipyridine system produced cis-products, demonstrating a remarkable control over the stereochemical outcome. However, upon subjecting butyl-substituted enyne to the standard reaction conditions, the desired products were not obtained. Intriguingly, the utilization of both Mn(III)- and Cu(II)-mediated reactions with N-alkyl substrates unexpectedly led to the exclusive formation of cis- products. It is hypothesized that the presence of electron- donating alkyl substituents alters the inherent characteristics of the substrates, particularly by reducing their steric hindrance. This diminished steric bulk potentially renders the cyclized rings overly flexible, impeding the efficient coordination of the manganese complex with the nitrogen atoms. Consequently, this structural feature results in a uniform selectivity, as observed experimentally (Scheme 6a). Upon introducing a mixture comprising LiCl, Mn(OAc)3•2H2O, and TMSN3 into the reaction system, the formation of a chloro-substituted product was unambiguously observed. This observation directly substantiates the hypothesis that an Mn(III)-Cl complex is initially formed and subsequently undergoes a chloro-ligand transfer oxidation process, ultimately yielding the chloro-substituted product. Consequently, they believed that an LnM-N3 species is formed in situ and subsequently participates in the diastereoselectivity-determining step (Scheme 6b).
2.2 Formation of six/S-five-membered ring
Sulfur-containing heterocyclic compounds hold a central role in organic chemistry, with extensive applications in pharmaceuticals, agrochemicals, natural products, and functional materials. Consequently, the development of sustainable and efficient methodologies for constructing sulfur-heterocyclic frameworks remains a persistent pursuit in the synthetic community.[9] Li’s group[10] reported a novel copper-catalyzed cascade cyclization of 1,7-enynes with metal sulfides. This sulfur-incorporating approach offers a direct route to the crucial thiophene-fused quinolin-4(5H)-one framework, achieved through a sequential cyclization and double C—S bond formation cascade. Notably, the chemoselectivity of this 1,7-enyne cyclization, leading to either 1,3,3a,9b-tetrahydrothieno[3,4-c]quinolin-4(5H)-ones or 3,3a-dihydrothieno[3,4-c]quinolin-4(5H)-ones, can be tuned by adjusting the sulfur source. However, aliphatic alkynes remain unreactive in this process (Scheme 7).[10] The chemoselectivity exhibited towards the products was contingent upon the sulfur source, specifically Na2S•9H2O or K2S, which imply that the hydrogen atoms incorporated into the nascent C—H bonds might originate from H2O. To substantiate this hypothesis, control experiments employing D2O were conducted, the outcomes of which revealed that hydrogenation does not constitute sole step within the cyclization pathway. When 1,2-diphenylethyne was subjected to reaction with Na2S•9H2O under the optimized conditions, exclusively (E)-1,2-diphenylethene, a hydrogenated product, was obtained. This finding suggests that the addition of the sulfur atom occurs specifically at the acrylate moiety, further corroborating the proposed mechanism wherein the hydrogen source plays a pivotal role in shaping the product selectivity.
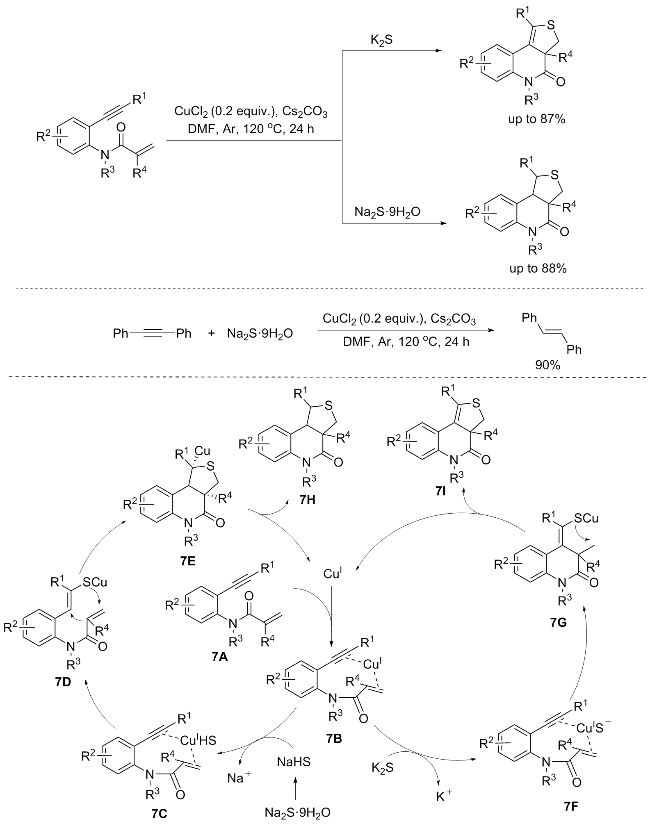
The mechanism depicted in Scheme 7 is proposed for the cascade cyclization reaction. Commencing with the coordination of the active CuI species to the C—C triple bond and the C—C double bond within the 1,7-enyne substrate 7A, intermediate 7B is formed. Subsequently, in the presence of Na2S•9H2O, intermediate 7B undergoes a rapid reaction with in situ generated NaHS, derived from the hydrolysis of Na2S•9H2O, to yield intermediate 7C. This is followed by an addition step, leading to the formation of intermediate 7D. Intermediate 7D then undergoes a cis- nucleophilic cyclization, resulting in the generation of intermediate 7E. The final step involves the protonation of intermediate 7E through trans-addition of water to the C—Cu bond, furnishing trans,trans-7H as the predominant isomer and simultaneously regenerating the active CuI species for further catalytic cycles. Alternatively, when K2S is employed as the sulfur source, intermediate 7B transforms into intermediate 7F. Subsequent nucleophilic addition and cyclization of intermediate 7F proceed to yield intermediate 7G. This is followed by a second nucleophilic cyclization event involving intermediate 7G, ultimately affording the product 7I.
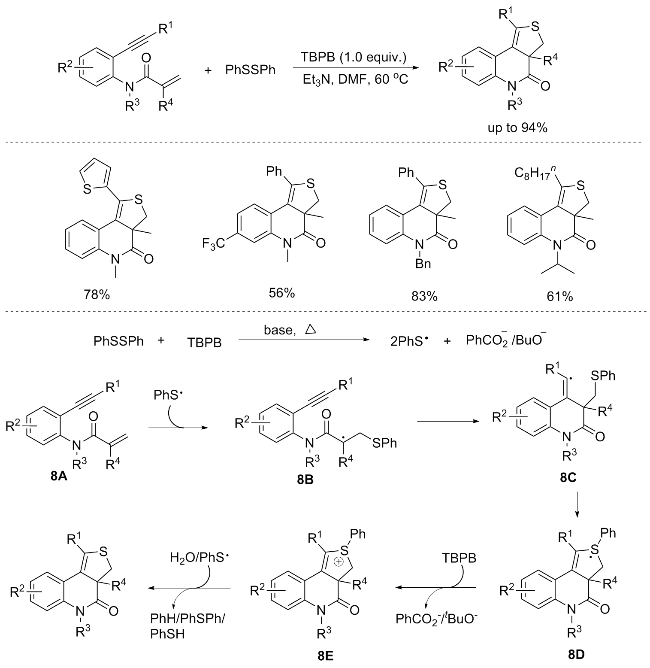
An innovative oxidative [2+2+1] heteroannulation of 1,7-enynes with disulfanes facilitated by Et3N, to yield 3,3-dihydro-thieno[3,4]quinolin-4(5H)-ones was reported by An’s group. This reaction, utilizing stoichiometric quan- tities of 1,7-enynes, sulfur atoms (disulfanes), tert-butyl peroxybenzoate (TBPB) as the oxidant, and Et3N as the base, provided unprecedented approach to the utilization of disulfanes as sulfur atom sources. Remarkably, enynes with an alkyl substituent at the terminal alkyne terminus are well compatible with this protocol. However, the attempted transformation of 2-benzyl-N-methyl-N-(2-(phenylethy- nyl)phenyl)acrylamide into the anticipated product proved unsuccessful under these conditions. It is noteworthy that sulfur-containing substrates, encompassing 1,2-bis(4-nitro- phenyl)disulfane, 1,2-diethyldisulfane, benzenethiol, diphenylsulfane, dimethyl sulfoxide, K2S, NaSCN, and S8, exhibited no discernible reactivity under the conditions. Furthermore, control experiments provide compelling evidence for a radical-mediated reaction pathway (Scheme 8).[11] The PhS· radical is efficiently generated via a single electron transfer (SET) process between PhSSPh and TBPB, facilitated by the presence of a base under thermal conditions. Subsequent addition of PhS• radical to the C=C bond of enyne 8A yields the alkyl radical intermediate 8B, which undergoes intramolecular cyclization to form the key intermediate 8C. This intermediate 8C then undergoes an annulation reaction, leading to the formation of intermediate 8D. Subsequently, intermediate 8D undergoes a single electron oxidation mediated by TBPB, accompanied by S—C(sp²) bond cleavage, ultimately furnishing the desired product.
Photocatalysis is a green chemistry technology that can convert light energy into chemical energy, thereby facilitating chemical reactions. Visible light-driven organic reactions have gained widespread attention and research in the past decade, significantly advancing the industrial application of these reactions.[12] In 2022, Wang’s group[13] found that, besides diphenyl disulfide (PhSSPh), diphenyl diselenide (PhSeSePh) also effectively reacted with N-(2- ethynylphenyl)acrylamides under photoirradiation. Remar-kably, S-heterocycles yield higher products than their Se analogs, likely due to selenium’s greater steric hindrance impeding the cyclization process. However, when the triple bond in N-(2-ethynylphenyl)acrylamides is terminal, the expected cyclic product is not formed. Additionally, diphenyl telluride ether was tested, but no reaction occurred. Attempts to scale up the reaction resulted in a significant yield decrease, suggesting that it may not be suitable for large-scale synthesis (Scheme 9).[13] Further mechanistic investigations were undertaken to elucidate the reaction pathway. Notably, the addition of TEMPO to the reaction mixture led to a pronounced suppression of the reaction, indicative of a radical-mediated process. Concurrently, under standard reaction conditions, the by-product PhSePh was unambiguously detected by high-resolution mass spectrometry (HRMS), providing additional evidence for the reaction mechanism. Furthermore, the reaction displayed no reactivity in the absence of light, underscoring the significance of the potential role of light in the radical cascade reaction.
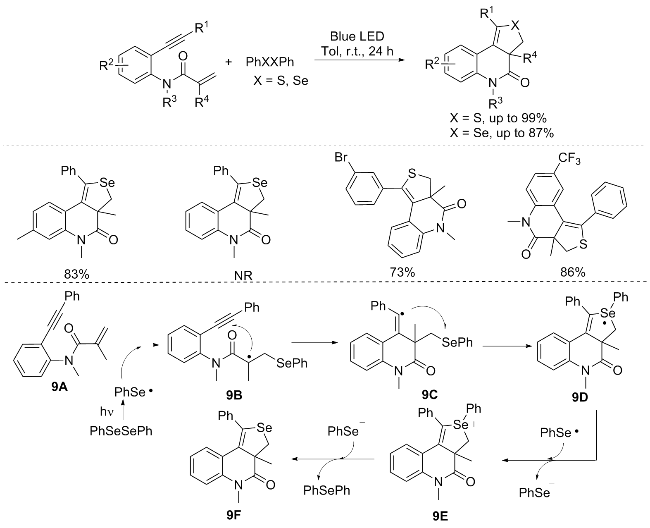
A plausible reaction mechanism is depicted in Scheme 9. Commencing with the assistance of blue light, the homolytic cleavage of diphenyl diselenide generates a selenium radical. This selenium radical subsequently attacks the C=C double bond of compound 9A, yielding the intermediate 9B. Following this, intermediate 9B undergoes an intramolecular annulation process, leading to the formation of the Se-heterocycle-fused polycyclic radical intermediate 9D. This intermediate 9D then sequentially undergoes a single-electron oxidation step, accompanied by the decomposition of the Se—C(sp²) bond, ultimately affording the desired product 9F alongside PhSePh as a by-product.
In 2013, Li’s group[14] introduced a novel metal-free oxidative [2+2+1] heteroannulation approach for the synthesis of thieno[3,4-c]quinolin-4(5H)ones from 1,7-enynes and thiocyanates. This transformation utilizes benzoyl peroxide (BPO) as the oxidant and sodium thiocyanate as the sulfur source, facilitating the formation of two C—S bonds and one C—C bond in a single-step process. This methodology offers a practical route to S-heterocycles while circumventing the need for metal catalysts and excessive bases. Notably, their attempts to extend this heteroannulation to aliphatic alkynes were unfruitful. Further investigation revealed that the addition of a radical scavenger effectively suppressed the reaction, indicating a radical-mediated mechanism. A plausible mechanism underlying the [2+2+1] heteroannulation reaction was described in Scheme 10. Commencing with the oxidation of KSCN by BPO, a single electron transfer event elicits the generation of the •SCN radical 10B. This radical subsequently attacks the C=C bond of the enyne substrate 10a, undergoing an addition reaction that prompts the formation of the vinyl radical intermediate 10D. Subsequent intramolecular cyclization of intermediate 10D fosters the emergence of the S-heterocycle-fused polycyclic radical intermediate 10E. This intermediate then undergoes a sequence of reactions, encompassing single electron oxidation steps, ultimately culminating in the decomposition of the S—CN bond. This transformation yields the final product, alongside PhCO2- CN as a by-product (Scheme 10).[14]
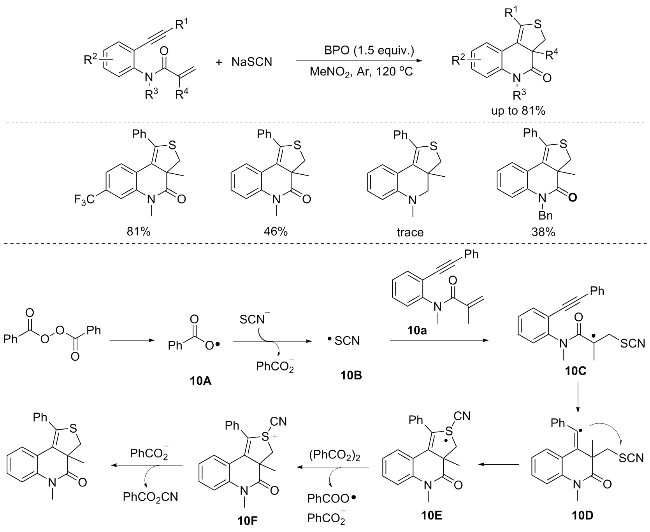
Utilizing N-(2-ethynylphenyl)acrylamides as substrates, the aforementioned reaction incorporates sulfur atoms into the five-membered ring of products via the employment of metal sulfides, disulfides, or thiocyanates as sulfur sources. These approaches offer practical strategy for the synthesis of thienoquinolinones and extends the synthetic scope of cyclopentaquinolinone derivatives. Notably, the incorporation of selenium atoms at the same position is also feasible, albeit with lower cycloaddition yields compared to sulfur. These strategies pave a novel pathway for the synthesis of diverse polycyclic heterocycles through various heteroatom incorporations.
2.3 Formation of six/O-five-membered ring
An ultrafast methodology for the synthesis of novel, highly functionalized 2-quinolinones from 1,7-enynes was realized by Andrade’s group.[15] This approach harnesses the power of microwave irradiation to synthesize 2-quino- linone-fused γ-lactones from Fentonʼs reagents in formamide. Through a sequence of six pivotal consecutive reactions, encompassing a diastereoselective step, the targeted 2-quinolinone-fused γ-lactones were obtained in commendable overall yields (up to 46%, 10 s). Notably, the synergistic effect of Fenton’s reagents (H2O2, FeSO4, and H2SO4) in formamide is pivotal in achieving this highly functionalized heterocycle. Furthermore, utilizing formamide as a solvent under microwave irradiation enabled the rapid formation of hydroxyl and carbamoyl radicals, leading to the swift synthesis of 2-quinolinone (Scheme 11).[15]
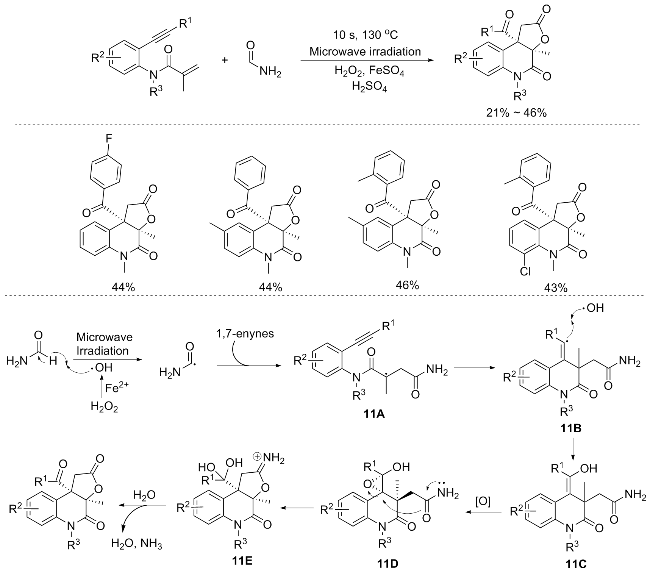
The proposed mechanism for the synthesis of 2-quino- linone-fused γ-lactones via the transformation of 1,7-enynes is outlined in Scheme 11. Initiation of the process involves the generation of hydroxyl radicals through a Fenton-like reaction, where H2O2 and Fe(II) in an acidic environment promote the formation of hydroxyl radicals, water, and Fe(III). Subsequently, Fe(II) is regenerated via the intera-ction of Fe(III) with H2O2. Formamide undergoes a radical abstraction by the hydroxyl radicals, yielding a carbamoyl radical. This carbamoyl radical then selectively attacks the carbon-carbon double bond of the acrylic amide segment, followed by a cyclization reaction with the adjacent carbon-carbon triple bond, resulting in the formation of vinyl radical 11B. The subsequent recombination of the hydroxyl radical with vinyl radical 11B furnishes enol intermediate 11C. Intermediate 11C, an enol species, undergoes an epoxidation reaction, transforming it into racemic epoxide 11D with the desired anti-stereochemistry. This configurational setup facilitates a 5-exo-tet cyclization, wherein the amide moiety acts as a nucleophile, attacking the less hindered side of the epoxide in an SN2-like fashion, affording intermediate 11E, a geminal diol. Finally, simple hydrolysis and dehydration of intermediate 11E lead to the formation of the desired product, accompanied by the elimination of ammonia.
3 Formation of six/six-membered ring
3.1 Formation of six/O-six-membered ring
In 2016, Li’s group[16] unveiled a novel iron-catalyzed radical [2+2+2] annulation strategy, wherein benzene- linked 1,7-enynes underwent efficient and selective reactions with aldehydes to yield a diverse array of fused [6.6.6] pyran molecules. This aldehyde radical-mediated approach boasts a compelling dual functionality, initiating and subsequently terminating the domino cyclization in a single-pot procedure. However, it is noteworthy that aliphatic aldehydes and 1,7-enynes containing an internal olefin moiety were unsuccessful in yielding the desired product. Further investigations via control experiments indicated that the generation of radicals from the aldehydes serves as the initiating step for this [2+2+2] annulation reaction (Scheme 12).[16]
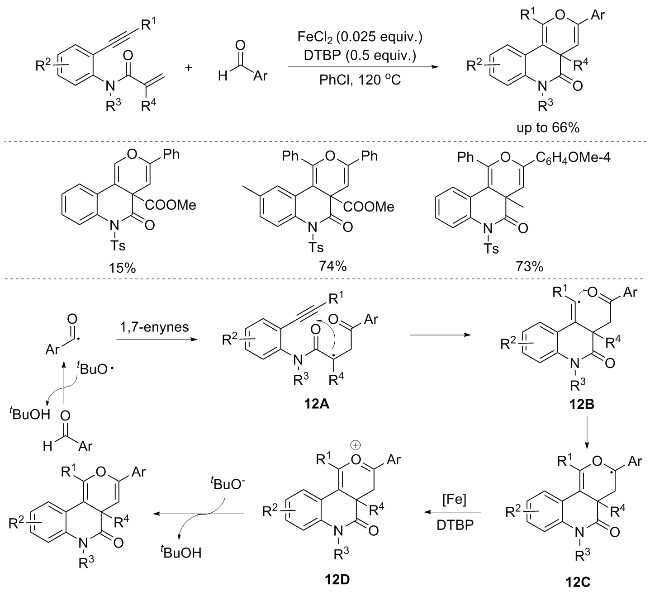
A plausible reaction pathway for the [2+2+2] annulation is depicted in Scheme 12. Initially, the C(sp²)—H bond within the aldehyde is cleaved by di-tert-butyl hydrogen peroxide (DTBP) under thermal conditions, yielding the acyl radical. Subsequently, this acyl radical undergoes an addition across the C—C double bond of the enyne, resulting in the formation of radical intermediate 12A. Subsequently, 12A undergoes cyclization with the adjacent C—C triple bond, affording vinyl radical intermediate 12B. Intermediate 12B then participates in a 6-endo-trig cyclization with the carbonyl group, leading to the generation of intermediate 12C. Subsequently, an iron-catalyzed irreversible oxidation of 12C occurs, yielding the oxonium cation 12D. Finally, deprotonation of 12D results in the formation of the desired annulation product.
In 2016, Xu’s group[17] introduced an innovative visible- light-mediated cascade cyclization strategy, involving the coupling of benzene-linked 1,7-enynes with benzoyl chlo-rides, to synthesize a diverse array of fused pyran compounds. This methodology offers a novel route for the generation of acyl radicals from benzoyl chlorides, thereby enabling an efficient synthesis of fused pyran molecules. However, alkylalkynes, including tert-butylalkyne and cyclopropylalkyne, remained inert under the reaction conditions, failing to yield the desired product. The Stern- Volmer analysis unveiled a noteworthy quenching effect of benzoyl chloride on the photoluminescence of fac-Ir(ppy)3 in acetonitrile, underscoring the specific interaction between the two substrates. Conversely, 2,6-lutidine, when presents in the same solution, does not exhibit a quenching effect on the excited state of fac-Ir(ppy)3, emphasizing the selectivity of the interaction. To further probe the nature of the reaction mechanism, a radical-trapping experiment was conducted, revealing that the addition of TEMPO, completely abolished the quenching reaction. This decisive observation demonstrates that the observed quenching is indeed mediated by the acyl radical generated from benzoyl chloride, thereby validating the initiation of a radical reaction pathway (Scheme 13).[17]
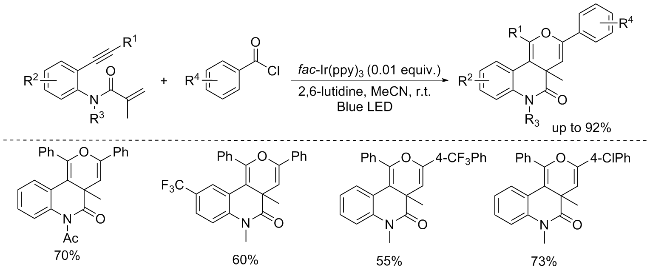
In 2023, Liu’s group[18] unveiled an innovative visible- light-driven strategy for the generation of aryl acyl radicals from readily accessible triazine esters. This methodology facilitates the synthesis of the desired product by coupling N-methyl-N-(2-ethynylphenyl)methacrylamide with triazine esters. Notably, the by-product 2-hydroxy-4,6-dimeth- oxy-1,3,5-triazine (HO-NMT) generated during the acylation reaction can be efficiently recycled and converted back to the corresponding triazine esters. This environmentally benign and versatile approach holds significant promise as a platform for the preparation of more intricate carbonyl- bearing molecules (Scheme 14).[18]
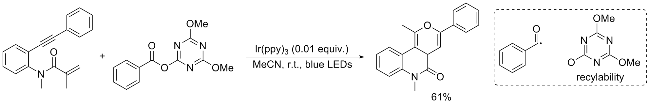
3.2 Formation of six/Si-six-membered ring
Organosilicon compounds play an important role in various aspects of contemporary chemistry, showcasing a remarkable versatility. Notably, organosilanes serve as indispensable reagents in chemical synthesis, particularly in reactions like the Fleming-Tamao, Hosomi-Sakurai, and Hiyama couplings, which underscore their pivotal role in molecular construction.[19] Additionally, the strategic incorporation of silicon into pharmaceutical molecules has been shown to enhance their potency and optimize pharmacokinetics, thereby broadening their therapeutic potential.[20] Beyond these applications, organosilanes have garnered significant attention as components in lubricants, adhesives, and a diverse array of advanced polymer materials, underscoring their widespread utility.[21] Consequently, the pursuit of novel and efficient methodologies for the synthesis of organosilanes continues to be a highly desirable in the field of organic chemistry.
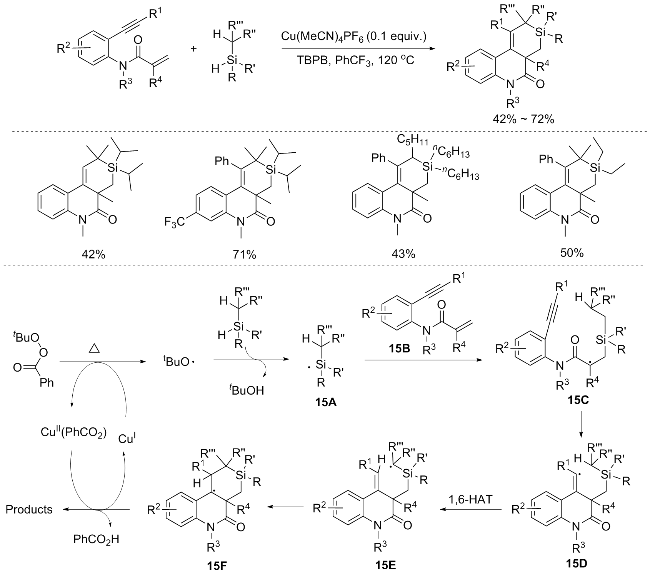
He’s group[22] devised a copper-catalyzed oxidative radical methodology that circumvents the use of costly noble metal/ligand catalytic systems. This approach selectively activates dual chemical bonds centered around the Si-atom, exploiting the unique characteristics of alkylsilanes. In the case of tertiary silanes, intermolecular oxidative annulation cascades with N-(2-(ethynyl)aryl)acrylamides lead to selective functionalization of Si—H/silyl C(sp3)—H bonds, affording silino[3,4-c]quinolin-5(3H)-ones. Conversely, with secondary silanes and HSi(TMS)3, either dual Si—H bonds or Si—H/Si—Si bonds are selectively cleaved, resulting in the formation of 4H-silolo[3,4-c]quinolin-4-ones. Notably, aliphatic alkynes also exhibit commendable reactivity within this system.
The proposed mechanisms for the reaction is elabo-rated in Scheme 15. In the presence of active CuI species, TBPB undergoes thermal decomposition, readily yielding the tert-butoxy radical alongside the CuII(PhCO2) complex. This tert-butoxy radical subsequently attacks the Si—H bond of tertiary silanes, leading to the generation of silicon-centered radical 15A. Subsequent addition of 15A across the C=C bond of the substrate 15B results in the formation of alkyl radical intermediate 15C. The intermediate 15C undergoes annulation, yielding the vinyl radical intermediate 15D, which then participates in a 1,6-hydrogen atom transfer (HAT) process with the silyl C(sp³)—H bond. This transfer gives the silyl alkyl radical intermediate 15E, which further undergoes annulation to produce intermediate 15F. Finally, the oxidation of intermediate 15F, mediated by the CuII(PhCO2) species via a single electron transfer, followed by deprotonation, culminates in the formation of the final product (Scheme 15).[22]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4 Conclusions and outlook
The tandem cyclization of N-(2-ethynylphenyl)acryl-amides has emerged as a versatile platform for rapid access to a variety of complex cyclic compounds. Beyond their atom economy, high cyclization efficiency, and exceptional functional group tolerance, these reactions circumvent the need for pre-functionalization of reaction substrates. The azide radical-initiated cyclization of N-(2-ethynylphenyl)-acrylamides has been scarcely reported, partly due to the challenges in controlling diverse radicals during prolonged one-pot operations, the inherent difficulty in generating three-membered rings, and the poor selectivity of radical addition towards alkenyl and alkynyl moieties.
Instances of transition metal-catalyzed N-(2-ethynyl-phenyl)acrylamide cyclization, particularly those leading to the synthesis of six-membered chiral heterocyclic compounds via corresponding asymmetric reactions, are exceedingly rare. This scarcity may stem from the fact that, in metal-catalyzed alkyne cyclization reactions, the formation of a seven-membered 1,7-alkyne-metal chelation ring poses greater difficulties compared to the formation of a six-membered 1,6-alkyne-metal chelation ring. Conversely, 1,7-enyl radical cyclization/cascade reactions occur readily due to the formation of more stable six-membered cyclic radical intermediates; nonetheless, the stereochemical outcomes of these radical cyclization reactions are difficult to control. At present, the formation of six/four-membered ring examples remains unreported, marking an intriguing direction worthy of investigation. In most cases, aliphatic alkynes are not suitable, this problem also deserves our further study. Additionally, the asymmetric reactions invol- ving N-(2-ethynylphenyl)acrylamides represent a captivating research frontier.
(Lu, Y.)