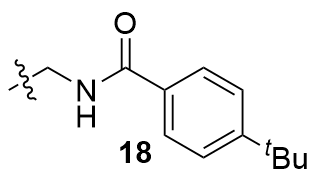
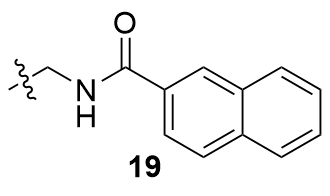
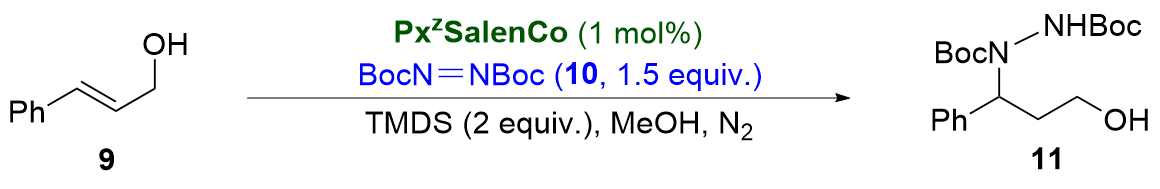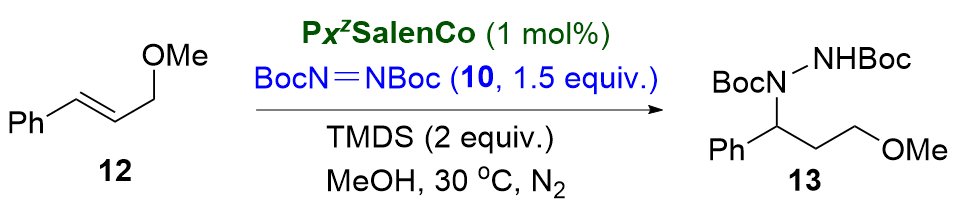Recently, great advances have been made in hydrofunctionalization of alkenes and alkynes via first row transition metal catalysis.
[13] In this context, the catalytic activity of our peptidic Salen cobalt complexes was evaluated with the model reaction of cobalt-catalyzed hydrohydrazination of cinnamic alcohol
9 with azodicarboxylate
10.
[14] We envisioned that the hydroxyl group in olefin
9 could engage in hydrogen-bonding interactions with the peptide on the cobalt Salen complex. The reaction was carried out in MeOH under N
2 at 30 ℃ with excess tetramethyldisiloxane (TMDS) as the hydride source. The results are shown in
Table 1. Although 1 mol%
P11SalenCo was ineffective, the same amount of complexes
P12SalenCo and
P13SalenCo exhibited excellent catalytic activity, resulting in clean conversion (in 3 h) of allylic alcohol
9 to 1,3-hydroxyl- alkylhydrazine
11 in 81% and 90% isolated yields, respectively (Entry 1 vs Entries 2, 3). With
P2 modified Salen- Co(II) complexes, a similar trend was still observed, even lower yields were obtained (Entry 4 vs Entries 5, 6).
P32SalenCo, carrying highly hydrophobic pentapeptide
P3, was effective to promote the reaction as well to give
11 almost quantitatively (Entry 7). Both heptapeptide
P4 modified cobalt complexes
P42SalenCo and
P43SalenCo could catalyze this hydrohydrazination at 30 ℃, affording
11 in excellent yields (Entries 8, 9), even though the former needed more time to achieve full conversion. At 15 ℃, the catalytic efficiency for all the peptidic cobalt complexes derived from framework
SA2 decreased remarkably (Entries 10~13). When quenched at 3 h, an 80% conversion was realized with tetrapeptide
P1 modified
P12SalenCo as the catalyst. Similarly, with complexes
P22SalenCo,
P32SalenCo and
P42SalenCo, 60%, 40% and 50% conversion were obtained respectively in the same reaction time, which roughly correlated proportionally with the reaction rates. The rate differences related to the decorating peptides are recognizable and should not be ignored. In order to make further comparison, non-peptide Salen-Co complexes
1SalenCo~
3SalenCo were also tested with the same reaction. Surprisingly,
1SalenCo displayed much higher catalytic activity than
P11SalenCo and
P21SalenCo, and the reaction could proceed to completion in 3 h even at 15 ℃ (Entry 14 vs Entries 1, 4). In contrast, both
2SalenCo and
3SalenCo were able to achieve only partial conversion with the same reaction time and temperature, showing slightly decreased catalytic efficiency than their peptidic congeners (Entries 15, 16 vs Entries 10~13). These preliminary data clearly manifest that not only the electronic nature of the Salen core is strongly correlated to the catalytic activity of the metal centre, but also the identity of the peptide can impose a significant influence, a feature reminiscent of metal cofactors in metalloenzymes. Although the exact reason remains elusive at this stage, the temperature effects on these catalytic reactions may reflect the common principle that peptides have lower conformational flexibility and hydrogen bonding strength at lower temperatures.



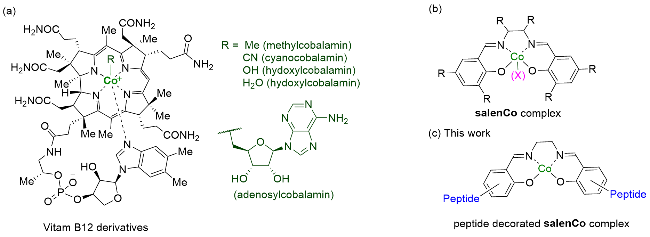
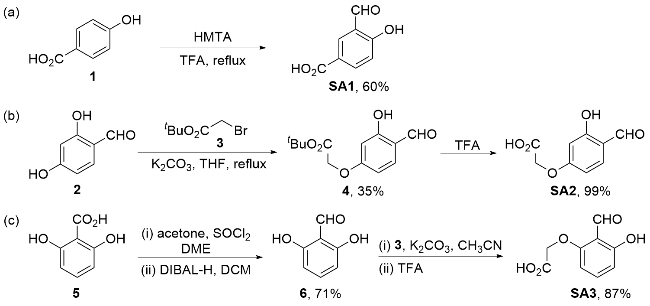
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}