化学学报 ›› 2021, Vol. 79 ›› Issue (4): 472-480.DOI: 10.6023/A20100489 上一篇 下一篇
所属专题: 纪念南开大学化学学科创建100周年
综述
付浩浩a, 陈淏川a, 张宏a, 邵学广a,b,*( ), 蔡文生a,*()
), 蔡文生a,*()
投稿日期:2020-10-24
发布日期:2020-12-02
通讯作者:
邵学广, 蔡文生
作者简介: |
付浩浩, 本科和博士均毕业于南开大学化学系. 现为南开大学博士后、助理研究员. 研究方向为增强采样算法开发、蛋白质-配体准确结合自由能计算策略研究和复杂体系中高度耦合的运动研究. |
 |
邵学广, 南开大学教授, 博士生导师, 1992年获中国科学技术大学中日联合培养博士学位. 2002 年获教育部第三届高校青年教师奖, 2003 年获国家自然科学基金委杰出青年基金. 主要从事化学计量学及近红外光谱分析方面的研究工作. 建立了小波变换和免疫算法用于复杂信号解析和在线处理的新方法以及一系列用于近红外光谱信号处理和建模的化学计量学方法. |
 |
蔡文生, 南开大学教授, 博士生导师. 1994年获中国科学技术大学博士学位. 主要从事分子模拟与理论化学计算领域的研究工作, 包括优化算法、自由能计算方法、分子模拟及理论化学计算在蛋白质-配体、药物载体、分子机器中的应用研究. |
基金资助:
Haohao Fua, Haochuan Chena, Hong Zhanga, Xueguang Shaoa,b,*(), Wensheng Caia,*()
Received:2020-10-24
Published:2020-12-02
Contact:
Xueguang Shao, Wensheng Cai
About author:Supported by:文章分享
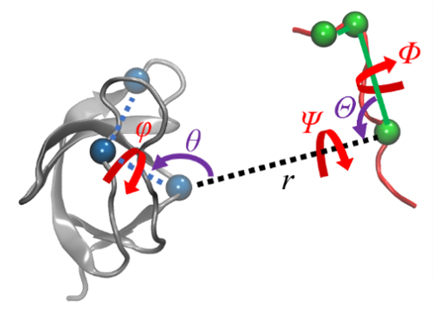
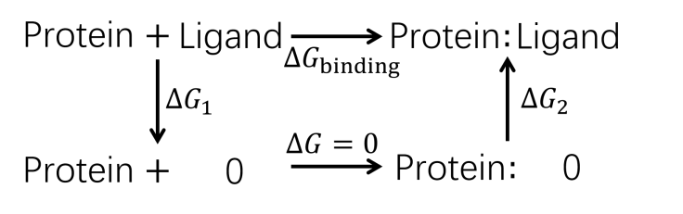
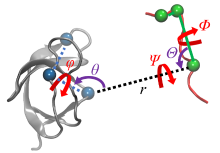
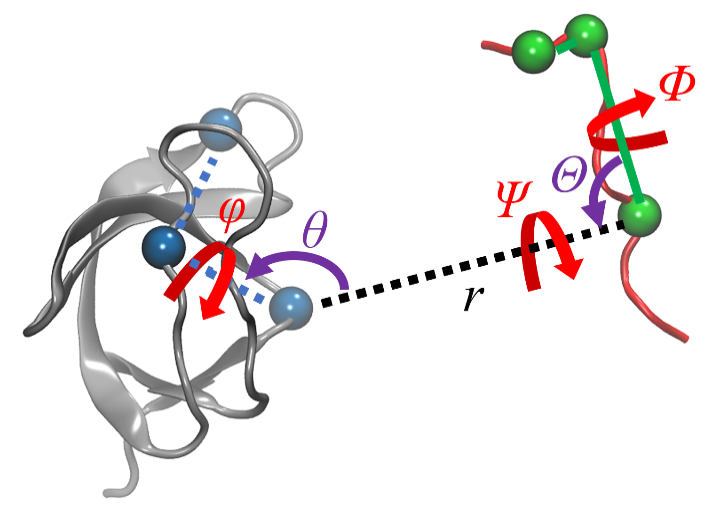
蛋白质-配体的结合过程伴随着复杂的结构变化, 在分子模拟可及的时间尺度内难以完全捕获, 这使得准确估计蛋白质-配体的结合自由能十分困难. 一种有效的解决途径是采用几何约束减小需要采样的构象空间, 再通过后处理方式扣除约束的影响. 本文综述了三种几何约束策略——漏斗状约束、球形约束和七自由度约束与自由能计算算法结合准确计算结合自由能的原理和进展, 重点概述理论严谨的七自由度约束的最新进展以及与Alchemistry或重要性采样方法的联用策略, 最后, 讨论了如何针对不同体系选择合适的计算策略以及蛋白质-配体准确结合自由能计算在药物设计等领域中的挑战和前景, 并提出了将上述方法进一步运用于研究更复杂的蛋白质-蛋白质问题的可能性.
付浩浩, 陈淏川, 张宏, 邵学广, 蔡文生. 基于几何约束的蛋白质-配体准确结合自由能计算[J]. 化学学报, 2021, 79(4): 472-480.
Haohao Fu, Haochuan Chen, Hong Zhang, Xueguang Shao, Wensheng Cai. Accurate Estimation of Protein-ligand Binding Free Energies Based on Geometric Restraints[J]. Acta Chimica Sinica, 2021, 79(4): 472-480.



| 步骤 | 对应项 | 集合变量 | 约束 | 体系 |
|---|---|---|---|---|
| 1 | RMSD | - | 蛋白质-配体 | |
| 2 | RMSD | 蛋白质-配体 | ||
| 3 | RMSD, | 蛋白质-配体 | ||
| 4 | RMSD, , | 蛋白质-配体 | ||
| 5 | RMSD, , , | 蛋白质-配体 | ||
| 6 | RMSD, , , , | 蛋白质-配体 | ||
| 7 | RMSD, , , , , | 蛋白质-配体 | ||
| 8 | RMSD | - | 配体 | |
| 9 | - | - | 解析计算 |
| 步骤 | 对应项 | 集合变量 | 约束 | 体系 |
|---|---|---|---|---|
| 1 | RMSD | - | 蛋白质-配体 | |
| 2 | RMSD | 蛋白质-配体 | ||
| 3 | RMSD, | 蛋白质-配体 | ||
| 4 | RMSD, , | 蛋白质-配体 | ||
| 5 | RMSD, , , | 蛋白质-配体 | ||
| 6 | RMSD, , , , | 蛋白质-配体 | ||
| 7 | RMSD, , , , , | 蛋白质-配体 | ||
| 8 | RMSD | - | 配体 | |
| 9 | - | - | 解析计算 |
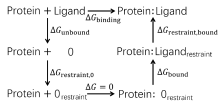
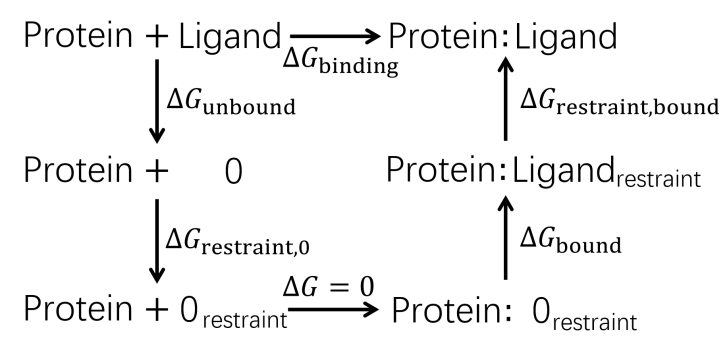
| 步骤 | 对应项 | 消失/生长部分 | 体系 |
|---|---|---|---|
| 1 | 配体 | 蛋白质-配体 | |
| 2 | 配体 | 配体 | |
| 3 | 七自由度约束 | 蛋白质-配体 | |
| 4 | (RMSD贡献) | RMSD约束 | 配体 |
| 5 | RMSD贡献以外部分) | 无 | 解析计算(式8) |
| 步骤 | 对应项 | 消失/生长部分 | 体系 |
|---|---|---|---|
| 1 | 配体 | 蛋白质-配体 | |
| 2 | 配体 | 配体 | |
| 3 | 七自由度约束 | 蛋白质-配体 | |
| 4 | (RMSD贡献) | RMSD约束 | 配体 |
| 5 | RMSD贡献以外部分) | 无 | 解析计算(式8) |
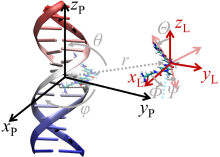
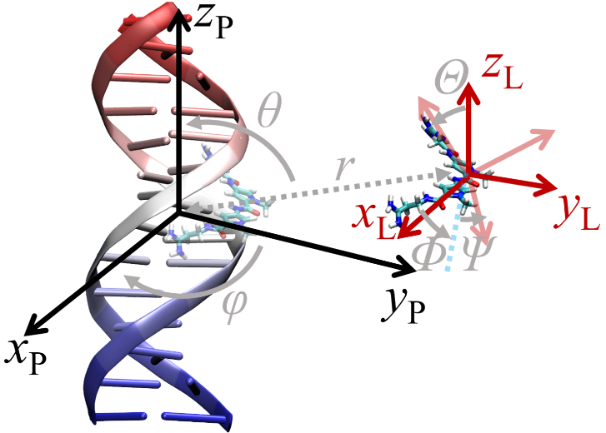
| 作者 | 使用策略 | 小分子配体个数 | 计算误差/ (kJ•mol–1) | 文献 |
|---|---|---|---|---|
| Aldeghi等 | alchemistry | 11 | MAE=2.5 | [ |
| Boyce等 | alchemistry | 13 | RMSE=7.5 | [ |
| Li等 | alchemistry | 100 | RMSE=2.59~6.44 | [ |
| Laury等 | alchemistry | 14 | MAE=5.02 | [ |
| Xie等 | alchemistry | 141 | RMSE=6.65 | [ |
| Deng等 | alchemistry/重要性采样 | 3 | AE=6.7~18 (alchemistry), 6.3~14 (重要性采样) | [ |
| Fu等 | 重要性采样 | 3(十肽) | AE<1.7 | [ |
| 作者 | 使用策略 | 小分子配体个数 | 计算误差/ (kJ•mol–1) | 文献 |
|---|---|---|---|---|
| Aldeghi等 | alchemistry | 11 | MAE=2.5 | [ |
| Boyce等 | alchemistry | 13 | RMSE=7.5 | [ |
| Li等 | alchemistry | 100 | RMSE=2.59~6.44 | [ |
| Laury等 | alchemistry | 14 | MAE=5.02 | [ |
| Xie等 | alchemistry | 141 | RMSE=6.65 | [ |
| Deng等 | alchemistry/重要性采样 | 3 | AE=6.7~18 (alchemistry), 6.3~14 (重要性采样) | [ |
| Fu等 | 重要性采样 | 3(十肽) | AE<1.7 | [ |
| 场景 | 建议 | 说明 |
|---|---|---|
| 基于数据库的药物筛选 | 分子对接 | 效率最高 |
| 结构相似配体结合能力的定性对比 | MM-G(P)BSA | 效率高, 使用简单 |
| 刚性小分子、结合位点在蛋白质表面 | 漏斗状约束或球状约束结合重要性采样方法 | 效率较高, 使用简单, 无需考虑小分子结构变化 |
| 刚性小分子、结合位点在蛋白质内部 | 基于六自由度约束的Alchemistry方法 | 无法使用重要性采样方法, 使用六自由度约束可以简单地解析计算约束贡献 |
| 柔性配体、结合位点在蛋白质表面 | 基于七自由度的重要性采样方法 | 同等条件下重要性采样算法收敛较快, 需要采用七自由度策略捕获柔性配体结构变化 |
| 柔性配体、结合位点在蛋白质内部 | 基于七自由度约束的Alchemistry方法 | 其他策略均不适合时最为“安全”的策略 |
| 场景 | 建议 | 说明 |
|---|---|---|
| 基于数据库的药物筛选 | 分子对接 | 效率最高 |
| 结构相似配体结合能力的定性对比 | MM-G(P)BSA | 效率高, 使用简单 |
| 刚性小分子、结合位点在蛋白质表面 | 漏斗状约束或球状约束结合重要性采样方法 | 效率较高, 使用简单, 无需考虑小分子结构变化 |
| 刚性小分子、结合位点在蛋白质内部 | 基于六自由度约束的Alchemistry方法 | 无法使用重要性采样方法, 使用六自由度约束可以简单地解析计算约束贡献 |
| 柔性配体、结合位点在蛋白质表面 | 基于七自由度的重要性采样方法 | 同等条件下重要性采样算法收敛较快, 需要采用七自由度策略捕获柔性配体结构变化 |
| 柔性配体、结合位点在蛋白质内部 | 基于七自由度约束的Alchemistry方法 | 其他策略均不适合时最为“安全”的策略 |
| [1] |
Chipot, C. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4,71.
|
| [2] |
Siebenmorgen, T.; Zacharias, M. WIREs Comput. Mol. Sci. 2019, 10,e1448.
|
| [3] |
de Ruiter A.; Oostenbrink C. Curr. Opin. Struct. Biol. 2020, 61,207.
|
| [4] |
Xu, J.; Wei, Y.; Wu, Z.; Yi, Z. Acta Chim. Sinica 2018, 76,408. (in Chinese)
|
|
( 徐婕, 魏雨晨, 伍智蔚, 易忠胜, 化学学报, 2018, 76,408.)
|
|
| [5] |
Cournia, Z.; Allen, B.K.; Beuming, T.; Pearlman, D.A.; Radak, B.K.; Sherman, W. J. Chem. Inf. Model. 2020, 60,4153.
|
| [6] |
Langan, R.A.; Boyken, S.E.; Ng, A.H.; Samson, J.A.; Dods, G.; Westbrook, A.M.; Nguyen, T.H.; Lajoie, M.J.; Chen, Z.; Berger, S.; Mulligan, V.K.; Dueber, J.E.; Novak, W.R. P.; El-Samad, H.; Baker, D. Nature 2019, 572,205.
|
| [7] |
Lemkul, J.A.; Huang, J.; Roux, B.; MacKerell, A.D. Chem. Rev. 2016, 116,4983.
|
| [8] |
Fan, Q.; Liang, H.; Xu, X.; Lv, S.; Liang, Z.; Yang, Y. Acta Chim. Sinica 2020, 78,547. (in Chinese)
|
|
( 范勤, 梁洪涛, 许贤祺, 吕松泰, 梁尊, 杨洋, 化学学报, 2020, 78,547.)
|
|
| [9] |
Nerenberg, P.S.; Head-Gordon, T. Curr. Opin. Struct. Biol. 2018, 49,129.
|
| [10] |
Chipot, C.; Pohorille, A. Free Energy Calculations, Springer, Berlin, Heidelberg, 2007.
|
| [11] |
Barducci, A.; Bonomi, M.; Parrinello, M. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1,826.
|
| [12] |
Laio, A.; Parrinello, M. Proc. Natl. Acad. Sci. 2002, 99,12562.
|
| [13] |
Fu, H.; Shao, X.; Cai, W.; Chipot, C. Acc. Chem. Res. 2019, 52,3254.
|
| [14] |
Fu, H.; Shao, X.; Chipot, C.; Cai, W. J. Chem. Theory Comput. 2016, 12,3506.
|
| [15] |
Chodera, J.D.; Mobley, D.L. Annu. Rev. Biophys. 2013, 42,121.
|
| [16] |
Zwanzig, R.W. J. Chem. Phys. 1954, 22,1420.
|
| [17] |
Kirkwood, J.G. J. Chem. Phys. 1935, 3,300.
|
| [18] |
Limongelli, V.; Bonomi, M.; Parrinello, M. Proc. Natl. Acad. Sci. 2013, 110,6358.
|
| [19] |
Raniolo, S.; Limongelli, V. Nat. Protoc. 2020, 15,2837.
|
| [20] |
Capelli, R.; Carloni, P.; Parrinello, M. J. Phys. Chem. Lett. 2019, 10,3495.
|
| [21] |
Boresch, S.; Tettinger, F.; Leitgeb, M.; Karplus, M. J. Phys. Chem. B 2003, 107,9535.
|
| [22] |
Woo, H.-J.; Roux, B. Proc. Natl. Acad. Sci. 2005, 102,6825.
|
| [23] |
Fu, H.; Cai, W.; Hénin, J.; Roux, B.; Chipot, C. J. Chem. Theory Comput. 2017, 13,5173.
|
| [24] |
Torrie, G.M.; Valleau, J.P. J. Comput. Phys. 1977, 23,187.
|
| [25] |
Kästner, J. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1,932.
|
| [26] |
Zong, Z.; Li, Q.; Hong, Z.; Fu, H.; Cai, W.; Chipot, C.; Jiang, H.; Zhang, D.; Chen, S.; Shao, X. J. Am. Chem. Soc. 2019, 141,14451.
|
| [27] |
Deng, N.; Cui, D.; Zhang, B.W.; Xia, J.; Cruz, J.; Levy, R. Phys. Chem. Chem. Phys. 2018, 20,17081.
|
| [28] |
Aldeghi, M.; Heifetz, A.; Bodkin, M.J.; Knapp, S.; Biggin, P.C. J. Am. Chem. Soc. 2017, 139,946.
|
| [29] |
Boyce, S.E.; Mobley, D.L.; Rocklin, G.J.; Graves, A.P.; Dill, K.A.; Shoichet, B.K. J. Mol. Biol. 2009, 394,747.
|
| [30] |
Li, Z.; Huang, Y.; Wu, Y.; Chen, J.; Wu, D.; Zhan, C.G.; Luo, H.B. J. Med. Chem. 2019, 62,2099.
|
| [31] |
Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. Comput. Phys. Commun. 2014, 185,604.
|
| [32] |
Fiorin, G.; Klein, M.L.; Hénin, J. Mol. Phys. 2013, 111,3345.
|
| [33] |
Sidky, H.; Colón, Y.J.; Helfferich, J.; Sikora, B.J.; Bezik, C.; Chu, W.; Giberti, F.; Guo, A.Z.; Jiang, X.; Lequieu, J.; Li, J.; Moller, J.; Quevillon, M.J.; Rahimi, M.; Ramezani-Dakhel, H.; Rathee, V.S.; Reid, D.R.; Sevgen, E.; Thapar, V.; Webb, M.A.; Whitmer, J.K.; de Pablo, J.J. J. Chem. Phys. 2018, 148,44104.
|
| [34] |
Ibrahim, P.; Clark, T. Curr. Opin. Struct. Biol. 2019, 55,129.
|
| [35] |
Wang, J.; Deng, Y.; Roux, B. Biophys. J. 2006, 91,2798.
|
| [36] |
de Ruiter, A.; Oostenbrink, C. Curr. Opin. Chem. Biol. 2011, 15,547.
|
| [37] |
Lee, T.-S.; Allen, B.K.; Giese, T.J.; Guo, Z.; Li, P.; Lin, C.; McGee, T.D.; Pearlman, D.A.; Radak, B.K.; Tao, Y.; Tsai, H.-C.; Xu, H.; Sherman, W.; York, D.M. J. Chem. Inf. Model. 2020, 60,5595.
|
| [38] |
Gumbart, J.C.; Roux, B.; Chipot, C. J. Chem. Theory Comput. 2013, 9,794.
|
| [39] |
Karplus, M.; Kuriyan, J. Proc. Natl. Acad. Sci. 2005, 102,6679.
|
| [40] |
Rizzi, A.; Grinaway, P.B.; Parton, D.L.; Shirts, M.R.; Wang, K.; Eastman, P.; Friedrichs, M.; Pande, V.S.; Branson, K.; Mobley, D.L.; Chodera, J.D. “YANK: A GPU-accelerated platform for alchemical free energy calculations,” can be found under getyank.org, 2020.
|
| [41] |
Mobley, D.L.; Chodera, J.D.; Dill, K.A. J. Chem. Theory Comput. 2007, 3,1231.
|
| [42] |
Henriksen, N.M.; Fenley, A.T.; Gilson, M.K. J. Chem. Theory Comput. 2015, 11,4377.
|
| [43] |
Heinzelmann, G.; Henriksen, N.M.; Gilson, M.K. J. Chem. Theory Comput. 2017, 13,3260.
|
| [44] |
Cruz, J.; Wickstrom, L.; Yang, D.; Gallicchio, E.; Deng, N. J. Chem. Theory Comput. 2020, 16,2803.
|
| [45] |
Coutsias, E.A.; Seok, C.; Dill, K.A. J. Comput. Chem. 2004, 25,1849.
|
| [46] |
Fu, H.; Gumbart, J.C.; Chen, H.; Shao, X.; Cai, W.; Chipot, C. J. Chem. Inf. Model. 2018, 58,556.
|
| [47] |
Xing, J.; Lu, W.; Liu, R.; Wang, Y.; Xie, Y.; Zhang, H.; Shi, Z.; Jiang, H.; Liu, Y.C.; Chen, K.; Jiang, H.; Luo, C.; Zheng, M. J. Chem. Inf. Model. 2017, 57,1677.
|
| [48] |
Wang, Y.; Li, L.; Zhang, B.; Xing, J.; Chen, S.; Wan, W.; Song, Y.; Jiang, H.; Jiang, H.; Luo, C.; Zheng, M. J. Med. Chem. 2017, 60,2026.
|
| [49] |
Zhang, X.; Li, X.; Wang, R. J. Chem. Inf. Model. 2009, 49,1033.
|
| [50] |
Pei, J.; Wang, Q.; Liu, Z.; Li, Q.; Yang, K.; Lai, L. Proteins Struct. Funct. Bioinforma. 2006, 62,934.
|
| [51] |
Liu, Y.; Xu, Z.; Yang, Z.; Chen, K.; Zhu, W. J. Mol. Model. 2013, 19,5015.
|
| [52] |
Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z. H.; Hou, T. Chem. Rev. 2019, 119,9478.
|
| [53] |
Ruiz-Blanco, Y.B.; Sanchez-Garcia, E. J. Chem. Theory Comput. 2020, 16,1396.
|
| [54] |
Aldeghi, M.; Heifetz, A.; Bodkin, M.J.; Knapp, S.; Biggin, P.C. Chem. Sci. 2016, 7,207.
|
| [55] |
Li, Z.; Li, X.; Huang, Y.-Y.; Wu, Y.; Liu, R.; Zhou, L.; Lin, Y.; Wu, D.; Zhang, L.; Liu, H.; Xu, X.; Yu, K.; Zhang, Y.; Cui, J.; Zhan, C.G.; Wang, X.; Luo, H.B. Proc. Natl. Acad. Sci. 2020, 117,27381.
|
| [56] |
Qian, Y.; Cabeza de Vaca, I.; Vilseck, J.Z.; Cole, D.J.; Tirado-Rives, J.; Jorgensen, W.L. J. Phys. Chem. B 2019, 123,8675.
|
| [57] |
Gumbart, J.C.; Roux, B.; Chipot, C. J. Chem. Theory Comput. 2013, 9,3789.
|
| [58] |
Rizzi, A.; Jensen, T.; Slochower, D.R.; Aldeghi, M.; Gapsys, V.; Ntekoumes, D.; Bosisio, S.; Papadourakis, M.; Henriksen, N.M.; de Groot, B.L.; Cournia, Z.; Dickson, A.; Michel, J.; Gilson, M.K.; Shirts, M.R.; Mobley, D.L.; Chodera, J.D. J. Comput. Aided. Mol. Des. 2020, 34,601.
|
| [59] |
Zhang, H.; Gattuso, H.; Dumont, E.; Cai, W.; Monari, A.; Chipot, C.; Dehez, F. Molecules 2018, 23,228.
|
| [60] |
Du, S.; Wang, H.; Yang, Y.; Feng, X.; Shao, X.; Chipot, C.; Cai, W. J. Phys. Chem. C 2019, 123,922.
|
| [61] |
Liu, H.; Fu, H.; Shao, X.; Cai, W.; Chipot, C. J. Chem. Theory Comput. 2020, 16,6397.
|
| [62] |
Laury, M.L.; Wang, Z.; Gordon, A.S.; Ponder, J.W. J. Comput. Aided. Mol. Des. 2018, 32,1087.
|
| [63] |
Xie, B.; Nguyen, T.H.; Minh, D.D. L. J. Chem. Theory Comput. 2017, 13,2930.
|
| [64] |
Jiang, W.; Roux, B. J. Chem. Theory Comput. 2010, 6,2559.
|
| [65] |
Jo, S.; Jiang, W. Comput. Phys. Commun. 2015, 197,304.
|
| [66] |
Miao, Y.; Feher, V.A.; McCammon, J.A. J. Chem. Theory Comput. 2015, 11,3584.
|
| [67] |
Yang, Y.I.; Niu, H.; Parrinello, M. J. Phys. Chem. Lett. 2018, 9,6426.
|
| [68] |
Fu, H.; Zhang, H.; Chen, H.; Shao, X.; Chipot, C.; Cai, W. J. Phys. Chem. Lett. 2018, 9,4738.
|
| [69] |
Fu, H.; Chen, H.; Wang, X.; Chai, H.; Shao, X.; Cai, W.; Chipot, C. J. Chem. Inf. Model. 2020, 60,5366.
|
| [70] |
Perthold, J.W.; Oostenbrink, C. J. Chem. Theory Comput. 2017, 13,5697.
|
| [71] |
Pan, A.C.; Sezer, D.; Roux, B. J. Phys. Chem. B 2008, 112,3432.
|
| [72] |
Suh, D.; Jo, S.; Jiang, W.; Chipot, C.; Roux, B. J. Chem. Theory Comput. 2019, 15,5829.
|
| [73] |
Brotzakis, Z.F.; Limongelli, V.; Parrinello, M. J. Chem. Theory Comput. 2019, 15,743.
|
| [1] | 程敏, 王诗慧, 罗磊, 周利, 毕可鑫, 戴一阳, 吉旭. 面向乙烷/乙烯分离的金属有机框架膜的大规模计算筛选[J]. 化学学报, 2022, 80(9): 1277-1288. |
| [2] | 王诗慧, 薛小雨, 程敏, 陈少臣, 刘冲, 周利, 毕可鑫, 吉旭. 机器学习与分子模拟协同的CH4/H2分离金属有机框架高通量计算筛选[J]. 化学学报, 2022, 80(5): 614-624. |
| [3] | 杨磊, 吴宇静, 吴选军, 蔡卫权. 面向C4烯烃混合物吸附分离的真实金属-有机骨架材料高通量筛选[J]. 化学学报, 2021, 79(4): 520-529. |
| [4] | 蔡铖智, 李丽凤, 邓小梅, 李树华, 梁红, 乔智威. 基于机器学习和高通量计算筛选金属有机框架的甲烷/乙烷/丙烷分离性能[J]. 化学学报, 2020, 78(5): 427-436. |
| [5] | 刘治鲁, 李炜, 刘昊, 庄旭东, 李松. 金属有机骨架的高通量计算筛选研究进展[J]. 化学学报, 2019, 77(4): 323-339. |
| [6] | 卞磊, 李炜, 魏振振, 刘晓威, 李松. 基于高通量计算筛选的金属有机骨架材料甲醛吸附性能[J]. 化学学报, 2018, 76(4): 303-310. |
| [7] | 杨文远, 梁红, 乔智威. 高通量筛选金属-有机框架:分离天然气中的硫化氢和二氧化碳[J]. 化学学报, 2018, 76(10): 785-792. |
| [8] | 刘蓓, 廉源会, 李智, 陈光进. 生物金属-有机骨架材料中药物吸附及扩散的分子模拟研究[J]. 化学学报, 2014, 72(8): 942-948. |
| [9] | 刘蓓, 唐李兴, 廉源会, 李智, 孙长宇, 陈光进. 互穿结构及混合配体对金属-有机骨架材料分离性能的影响[J]. 化学学报, 2013, 71(06): 920-928. |
| [10] | 陈晓光, 赵晓杰, 王嵩, 王丽萍, 李惟, 孙家钟. Sirt1及Sirt2与活性分子INA的作用机制研究[J]. 化学学报, 2013, 71(02): 199-204. |
| [11] | 李悦, 谷雨, 何佳, 何华, 周祎, Chuong Pham-Huy. 光谱法与分子模拟技术研究杨梅素与牛血清白蛋白的相互作用[J]. 化学学报, 2012, 70(02): 143-150. |
| [12] | 郑蓉, 吕暾. 毒素DON单链抗体的同源建模及与DON结合的分子模拟研究[J]. 化学学报, 2011, 69(23): 2882-2888. |
| [13] | 刘璐莎, 樊君, 胡春梅, 孙洋, 胡晓云, 赵英永, 魏嵩, 梁旭华. 土贝母皂苷II与人血清白蛋白相互作用机制的光谱研究[J]. 化学学报, 2011, 69(21): 2589-2596. |
| [14] | 徐飞, 张林群, 何立巍, 谷巍, 房方, 吴启南, 赵波. 泽泻醇类化合物与血清白蛋白相互作用的分子机理研究[J]. 化学学报, 2011, 69(19): 2228-2234. |
| [15] | 王公轲, 席辉, 田芳, 韩梦莹, 卢雁. 光谱和分子模拟法研究乙硫异烟胺与木瓜蛋白酶的分子作用机制[J]. 化学学报, 2011, 69(01): 95-100. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
