
有机-无机型杂多酸相转移催化氧化脱硫性能研究
收稿日期: 2014-08-29
网络出版日期: 2013-11-14
基金资助
项目受国家自然科学基金(Nos. 20976097,21076116,21211120165,21311120297);中国石油天然气集团公司石油科技创新基金(2010D-5006-0405)和山东省自然科学基金(ZR2011BM023)资助.
Study on Oxidative Desulfurization Catalyzed by Organic-inorganic Heteropolyacids as Phase Transfer Catalyst
Received date: 2014-08-29
Online published: 2013-11-14
Supported by
Project supported by the National Natural Science Foundation of China (Nos. 20976097, 21076116, 21211120165, 21311120297), Petro China Scientific and Technical Innovation Project (2010D-5006-0405) and Natural Science Foundation of Shandong Province (ZR2011BM023).
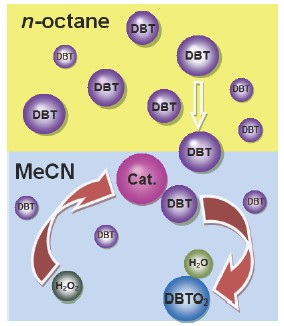
合成了四种有机-无机型杂多酸催化剂,包括[π-C5H5NC16H33]3[PW4O16],[π-C5H5NC16H33]3[PMo4O16],[π-C5H5NC12H25]3[PW4O16]和[π-C5H5NC12H25]3[PMo4O16]. 以有机硫的正辛烷溶液为模拟油品,H2O2为氧化剂,乙腈为萃取剂,在两相体系中,考察了上述四种催化剂对模拟油品中二苯并噻吩(DBT)氧化脱硫的催化活性. 结果表明,[π-C5H5NC16H33]3[PW4O16]具有最佳的催化活性. 采用[π-C5H5NC16H33]3[PW4O16]进行后续研究发现,反应完毕,[π-C5H5NC16H33]3[PW4O16]以沉淀的形式析出,可以重复利用且脱硫效果很好. 研究表明,上述有机-无机型杂多酸属于相转移催化剂,氧化脱硫反应体系属于反应控制相转移催化体系. 在相同实验条件下,由于电子云密度和空间位阻效应共同的作用,DBT、噻吩(TH)、苯并噻吩(BT)和4,6-二甲基二苯并噻吩(4,6-DMDBT)脱硫由易到难的顺序为DBT >4,6-DMDBT >BT >TH,并分别通过GC-MS分析确定它们的氧化产物. 将[π-C5H5NC16H33]3[PW4O16]进一步应用于柴油氧化脱硫,其中硫含量由355 mg/kg (mg/kg等同于ppmw)降至26 mg/kg,去除率达92.7%. 利用上述四种有机-无机型杂多酸作催化剂,研究DBT氧化反应过程动力学,确定DBT的表观反应级数均为一级,表观活化能为47.9~55.4 kJ/mol.
于凤丽 , 王睿 . 有机-无机型杂多酸相转移催化氧化脱硫性能研究[J]. 化学学报, 2014 , 72(1) : 105 -113 . DOI: 10.6023/A13080905
In this paper, organic-inorganic heteropolyacids were prepared, including [π-C5H5NC16H33]3[PW4O16], [π-C5H5NC16H33]3[PMo4O16], [π-C5H5NC12H25]3[PW4O16] and [π-C5H5NC12H25]3[PMo4O16]. The efficiency of oxidative desulfurization was investigated on the four kinds of catalysts under the same conditions using the simulated oil prepared by dissolving organo-sulfur in normal octane. The results show that the catalytic activity of [π-C5H5NC16 H33]3[PW4O16] is the best. The catalytic activity of these heteropolyacids was following the order of [π-C5H5NC16H33]3[PW4O16]>[π-C5H5NC12H25]3[PW4O16]>[π-C5H5NC16H33]3[PMo4O16]>[π-C5H5NC12H25]3[PMo4O16]. Under the optimal experimental conditions, pre-reaction time between H2O2 and the catalyst being 10 min, reaction temperature being 60 ℃, O/S molar ratio being 10, the amount of catalyst being 1 wt% of n-octane, and the catalyst being [π-C5H5NC16H33]3[PW4O16], the dibenzothiophene (DBT) conversion was nearly 100% after 60 min. The catalyst [π-C5H5NC16H33]3[PW4O16] can be reclaimed by auto precipitation due to the exhausting of H2O2. The results show that organic-inorganic heteropolyacids are phase transfer catalyst and oxidative desulfurization system belongs to the phase transfer catalysis system controlled by reaction. The catalytic activity of the recycled [π-C5H5NC16H33]3[PW4O16] is almost the same as the fresh. Then, the efficiency of oxidative desulfurization was investigated with different simulated oils using DBT, thiophene (TH), benzothiophene (BT) and 4,6-dimethyldibenzothiophene (4,6-DMDBT), respectively. Under the same conditions, the efficiencies of oxidative desulfurization decrease in the order of DBT>4,6-DMDBT>BT>TH, the result was influenced by electron density on the sulfur atoms and the steric hindrance. It showed that the final desulfurization product of TH, BT, DBT and 4,6-DMDBT was SO42-,SO42-, DBT sulphone and 4,6-DMDBT sulphone, respectively. [π-C5H5NC16H33]3[PW4O16] used in desulfurization of the real diesel and the sulfur removal rate was 92.7%. The sulfur content was reduced from 355 mg/kg to 26 mg/kg. Furthermore, by studying the kinetics of DBT oxidation by H2O2, the reaction order was found to be 1st to DBT. The activation energy ranged from 47.9 kJ/mol to 55.4 kJ/mol by different organic-inorganic heteropolyacids. It indicates the reaction process is quite fast in ODS. The new type of catalysts has a broad development prospect in oil desulfurization.
[1] Zannikos, F.; Lois, E.; Stournas, S. Fuel Process. Technol. 1995, 42, 35.
[2] Shiraishi, Y.; Taehibana, K.; Hirai, T.; Komasawa, I. Ind. Eng. Chem. Res. 2002, 41, 4362.
[3] Lü, F.-Z.; Zhan, F.-T.; Wang, P.; Yao, X.-B. Chem. Eng. Oil Gas 2006, 35, 114. (吕志凤, 战风涛, 王萍, 姚绪波, 石油与天然气化工, 2006, 35, 114.)
[4] Otsuki, S.; Nonaka, T.; Takashima, N. Energy Fuels 2000, 14, 1232.
[5] Murata, S.; Murata, K.; Kidena, K. Energy Fuels 2004, 18, 116.
[6] Zhou, X.; Li, J.; Wang, X.; Jin, K.; Ma, W. Fuel Process. Technol. 2009, 90, 317.
[7] Ma, X. L.; Zhou, A. N.; Song, C. S. Catal. Today 2007, 123, 276.
[8] Wang, B.; Zhou, J. P.; Ma, H. Z. J. Hazard. Mater. 2009, 164, 256.
[9] Wang, D.; Qian, E. W.; Amano, H.; Okata, K.; Ishihara, A.; Kabe, T. Appl. Catal. A: Gen. 2003, 253, 91.
[10] Stanger, K. J.; Angelici, R. J. Energy Fuels 2006, 20, 1757.
[11] Tang, X.-D.; Shui, L.-L.; Liu, L. Chin J. Catal. 2004, 25, 789. (唐晓东, 税蕾蕾, 刘亮, 催化学报, 2004, 25, 789.)
[12] Yazu, K.; Yamamoto, Y.; Furuya, T.; Miki, K.; Ukegawa, K. Energy Fuels 2001, 15, 1535.
[13] Zannikos, F.; Lois, E.; Stoumas, S. Fuel Process. Technol. 1995, 42, 35.
[14] Bonde, S. E.; Gore, W.; Dolbear, G. E.; Skov, E. R. Am. Chem. Soc., Div. Petrol. Chem. Prepr. 2000, 45, 364.
[15] Filippis, P. D.; Scarsella, M. Energy Fuels 2003, 17, 1452.
[16] Yu, G. X.; Lu, S. X.; Chen, H.; Zhu, Z. N. Energy Fuels 2005, 19, 447.
[17] Yu, G. X.; Lu, S. X.; Chen, H.; Zhu, Z. N. Carbon 2005, 43, 2285.
[18] Te, M.; Fairlbridge, C.; Ring, Z. Appl. Catal. A: Gen. 2001, 219, 267.
[19] Collins, F. M.; Lucy, A. R.; Sharp, C. J. Mol. Catal. A: Chem. 1997, 117, 397.
[20] Yazu, K.; Yamamoto, Y.; Furuya, T.; Miki, K.; Ukegawa, K. Energy Fuels 2001, 15, 1535.
[21] Yazu, K.; Furuya, T.; Miki, K.; Ukegawa, K. Chem. Lett. 2003, 32, 920.
[22] Trakarnpruk, W.; Rujiraworawut, K. Fuel Process. Technol. 2009, 90, 411.
[23] Wang, R.; Zhang, G. F.; Zhao, H. X. Catal. Today 2010, 149, 117.
[24] Zhang, M.; Zhu, W. S.; Xun, S. H.; Li, H. M.; Gu, Q. Q.; Zhao, Z.; Wang, Q. Chem. Eng. J. 2013, 220, 328.
[25] Wang, E. B.; Hu, C. W.; Xu, L. Introduction to Polyoxometalate Chemistry, Chemical Industry Press, Beijing, 1998, pp. 15~16. (王恩波, 胡长文, 许林, 多酸化学导论, 化学工业出版社, 北京, 1998, pp. 15~16.)
[26] Xi, Z. W.; Zhou, N.; Sun, Y.; Li, K. L. Science 2001, 292, 1139.
[27] Zhou, N.; Xi, Z.-W.; Cao, G.-Y.; Fan, S.-H. J. Mol. Catal. 2001, 15, 113. (周宁, 奚祖威, 曹国英, 范淑华, 分子催化, 2001, 15, 113.)
[28] Sun, Y.; Xi, Z. W.; Cao, G. Y. J. Mol. Catal. A: Chem. 2001, 166, 219.
[29] Zhou, N.; Xi, Z. W.; Cao, G. Y.; Gao, S. Appl. Catal. A: Gen. 2003, 250, 239.
[30] Salles, L.; Aubry, C.; Thouvenot, R.; Robert, F.; Doremieux-Morin, C.; Chottard, G.; Ledon, H.; Jeannin, Y.; Brégeault, J. M. Inorg. Chem. 1994, 33, 871.
[31] Salles, L.; Aubry, C.; Robert, F.; Chottard, G.; Thouvenot, R.; Ledon, H.; Brégeault, J. M. New J. Chem. 1993, 17, 367.
[32] Cavani, F. Catal. Today 1998, 41, 73.
[33] Zhang, G. F.; Wang, R.; Yu, F. L.; Zhao, H. X. Chem. Pap. 2009, 63, 617.
[34] Zhang, B. Y.; Jiang, Z. X.; Li, J.; Zhang, Y. N.; Lin, F.; Liu, Y.; Li, C. J. Catal. 2012, 287, 5.
[35] Chen, L. J.; Guo, S. H.; Zhao, D. S. Chinese J. Chem. Eng. 2006, 14, 835.
[36] Wang, R.; Yu, F. L.; Zhang, G. F.; Zhao, H. X. Catal. Today 2010, 150, 37.
[37] Li, F.-T.; Kou, C.-G.; Sun, Z.-M.; Hao, Y.-J.; Liu, R.-H.; Zhao, D.-S. J. Hazard. Mater. 2012, 205-206, 164.
[38] Zhang, M.; Zhu, W. S.; Xun, S. H.; Li, H. M.; Gu, Q. Q.; Zhao, Z.; Wang, Q. Chem. Eng. J. 2013, 220, 328.
[39] Zhu, W. S.; Huang, W. L.; Li, H. M.; Zhang, M.; Jiang, W.; Chen, G. Y.; Han, C. R. Fuel Process. Technol. 2011, 92, 1842.
/
| 〈 |
|
〉 |
