





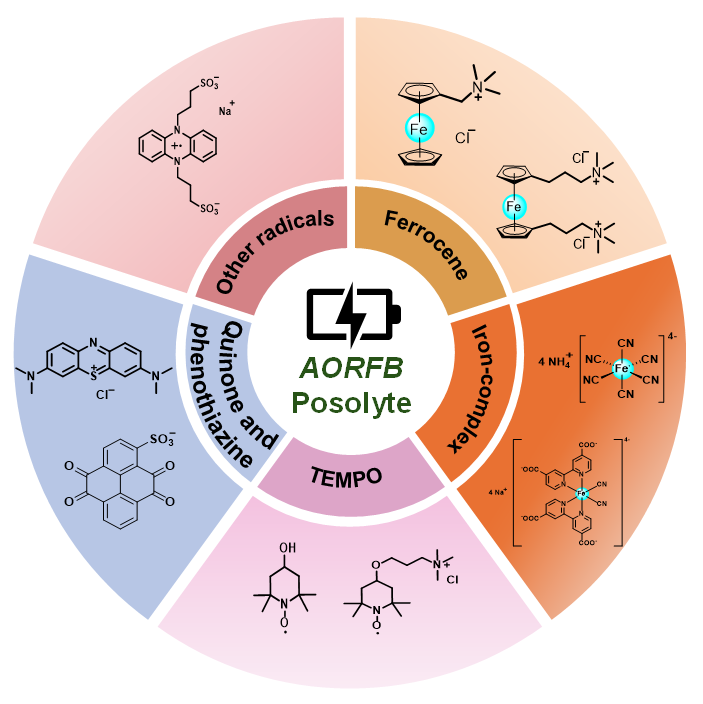
1 引言
1.1 液流电池背景

1.2 液流电池评价方式
1.3 有机储能活性材料的重要性质
1.3.1 氧化还原能力
1.3.2 水溶性
1.3.3 化学稳定性和电化学稳定性
1.4 研究现状

2 不同种类正极材料

2.1 二茂铁及其衍生物在水系有机液流电池中的应用
表1 二茂铁类衍生物作为正极储能活性材料应用于水系有机液流电池Table 1 Ferrocene derivatives used as posolyte in AORFBs |
| 年份 | 正极 | 负极 | 电压/V | 功率密度/ (mW•cm−2) | 能量效率/% | 容量密度/ (Ah•L−1) | 循环圈数/ 时间/d | 容量衰减率 每圈/每天/% | Ref. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2017 | 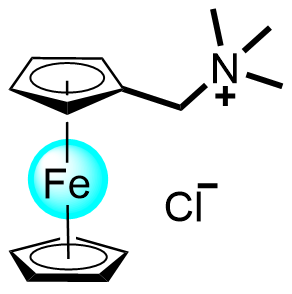 |  | 1.05 | 125 | 72 | 18.8 | 500/NA | 0.04/NA | [31] | ||||||||||
| 2017 | 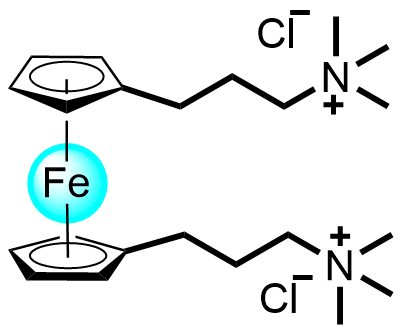 | 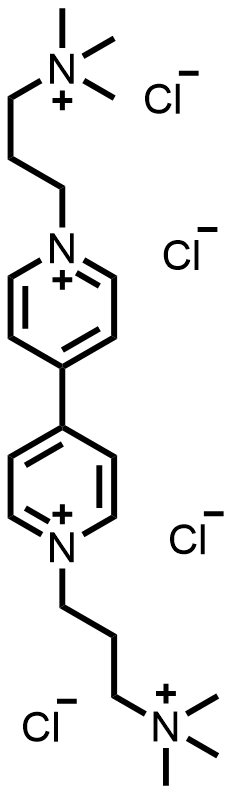 | 0.75 | 60 | 80 | 34.8 | 500/14 | 0.057/0.1 | [32] | ||||||||||
| 2020 | 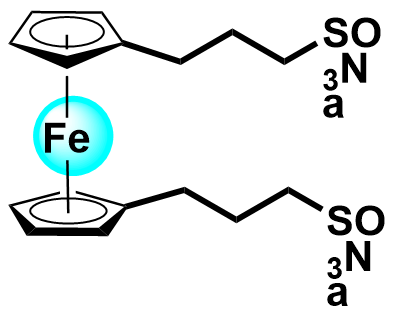 | Zn | 1.20 | NA | 83.3 | 40.2 | 1000/NA | 0.0025/NA | [33] | ||||||||||
| 2020 | 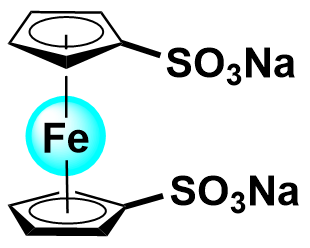 | 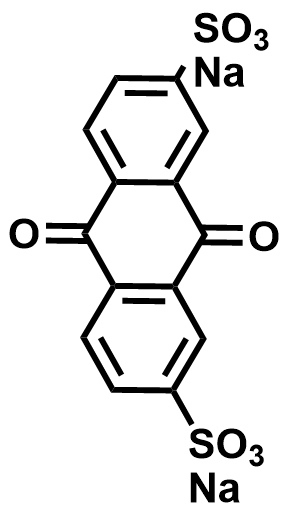 | 1.10 | NA | 80 | 8 | 100/NA | NA/NA | [34] | ||||||||||
| 2020 |  | 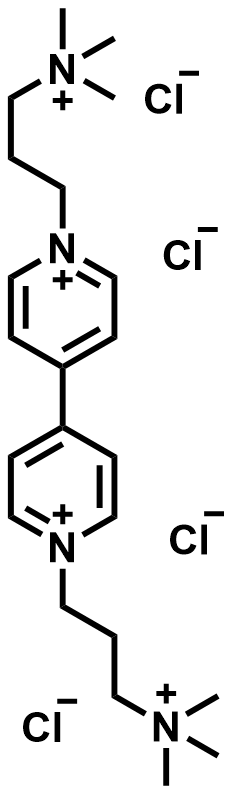 | 0.66 | 20.24 | 60 | 42 | 400/32 | NA/0.168 | [30] | ||||||||||
| 2021 | 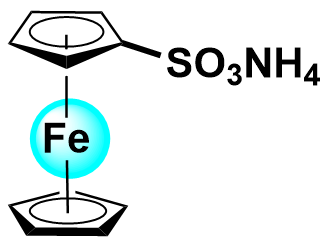 | ZnBr2 | 1.60 | NA | 83.2 | NA | 100/NA | 0.0745/NA | [35] | ||||||||||
| 2022 | 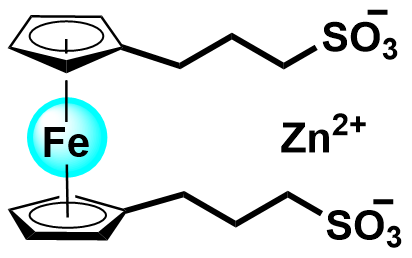 | 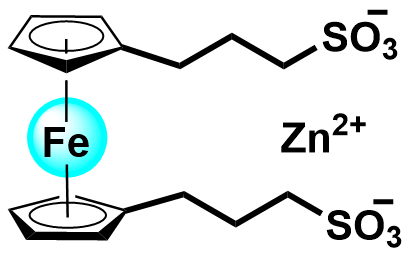 | 1.13 | 252.7 | 70 | 26.8 | 2000/53.5 | 0/0 | [36] |
“NA”, Not Available, 表示无相关数据报道. |

图4 A) FcNCl/MV液流电池示意图[31]; B) FcNCl储能活性材料降解机理; C) BTMAP-Vi与BTMAP-Fc化学结构; D) BTMAP-Vi与BTMAP-Fc循环伏安图[32]; E) BTMAP-Vi/BTMAP-Fc液流电池循环测试[32]; F)代表性的充电/放电容量电压图[32]; G)添加或不添加400 mg LiFePO4充电/放电容量电压图[33]Figure 4 A) Schematic representation of the FcNCl/MV AORFB[31]. Adapted with permission from [31] copyright 2017 Royal Society of Chemistry; B) Degradation mechanism of FcNCl; C) Chemical structure of the BTMAP-Vi/BTMAP-Fc; D) Cyclic voltammograms of BTMAP-Vi and BTMAP-Fc[32]; E) Cycling performance of the BTMAP-Vi/BTMAP-Fc AORFB[32]; F) Representative voltage vs capacity traces of selected cycles[32]. Adapted with permission from [32] copyright 2017 American Chemical Society; G) Voltage profiles of the flow battery with and without 400 mg LiFePO4 loaded in the posolyte tank[33]. Adapted with permission from [33] copyright 2020 Elsevier |
2.2 铁氰化物及其他铁配合物在水系有机液流电池中的应用
表2 铁氰化物及其他铁配合物作为正极储能活性材料应用于水系有机液流电池Table 2 Ferricyanide and other iron complex used as posolyte in AORFBs |
| 年份 | 正极 | 负极 | 电压/V | 功率密度/ (mW•cm−2) | 能量效率/% | 容量密度/ (Ah•L−1) | 循环圈数/ 时间/d | 容量衰减率 每圈/每天/% | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|
| 2019 |  | 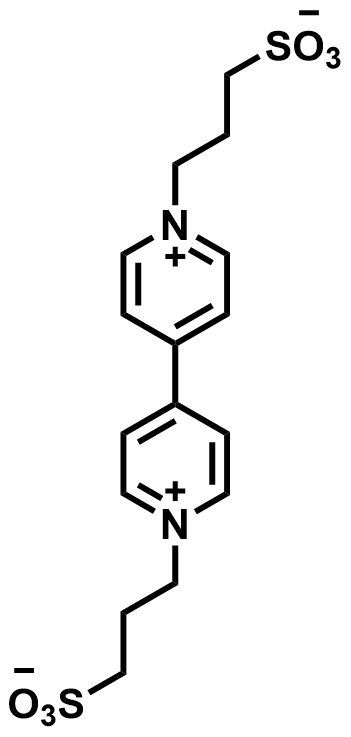 | 0.82 | 100 | 62.6 | 24.1 | 1000/45.8 | 0/0 | [38] | |
| 2020 | 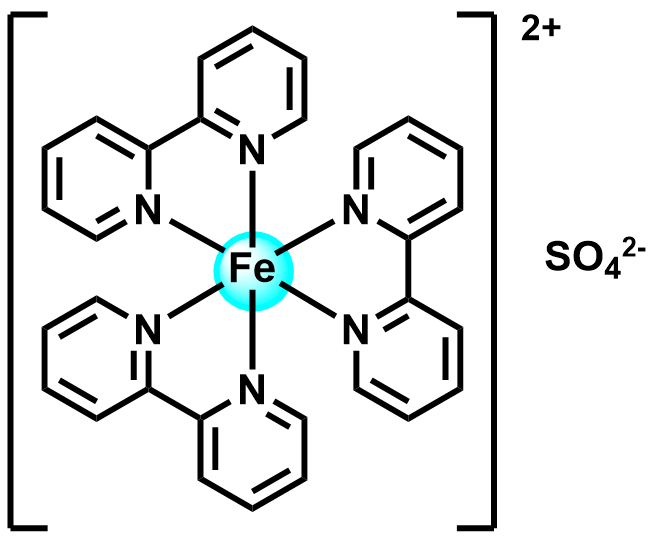 |  | 1.40 | 110 | 75.7 | NA | 215/NA | 0.06/NA | [39] | |
| 2021 | 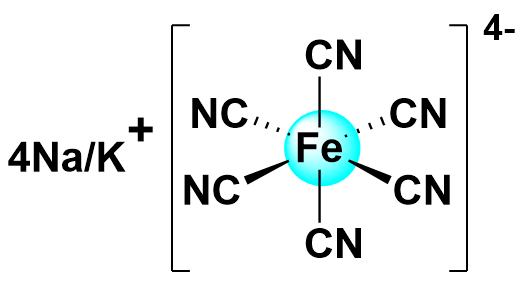 | K2S2 | 0.97 | 213.9 | 80.97 | 42.9 | 1500/30 | 0.002/NA | [40] | |
| 2021 | 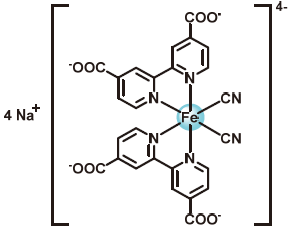 | 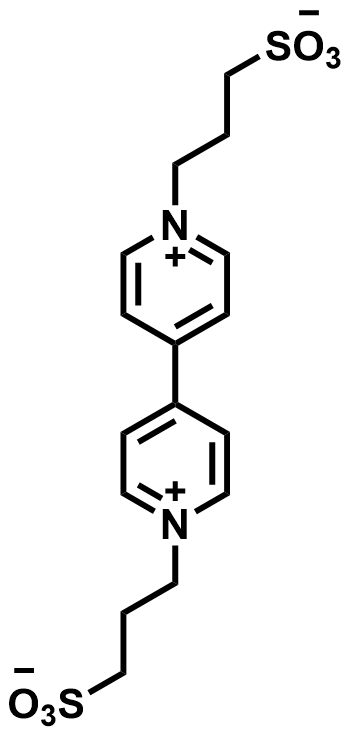 | 1.20 | NA | 70 | 26.8 | 6000/200 | 0.008/0.25 | [41] | |
| 2022 | 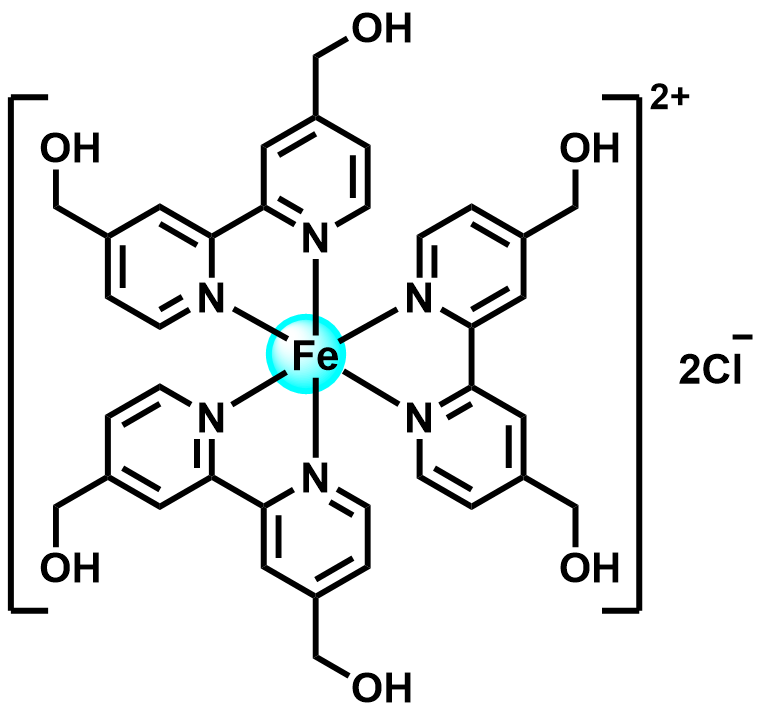 | 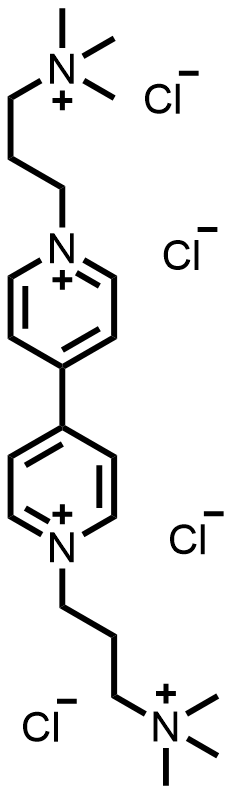 | 1.30 | 120 | 80 | 2.6 | 2700/25 | 0.0007/0.07 | [42] | |
| 2023 | 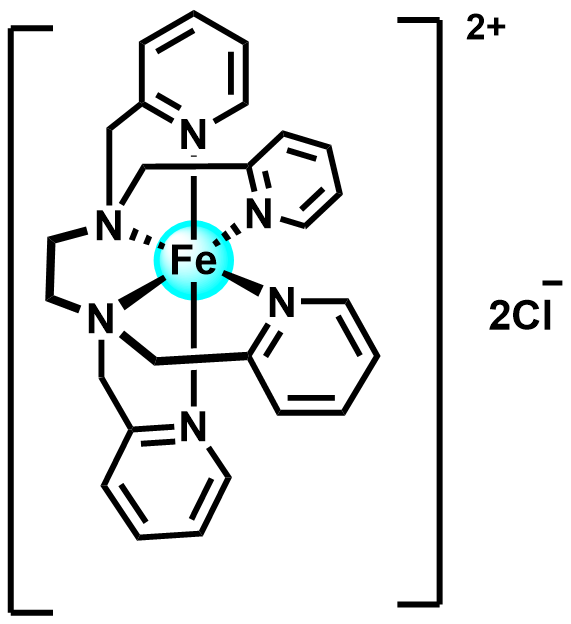 | 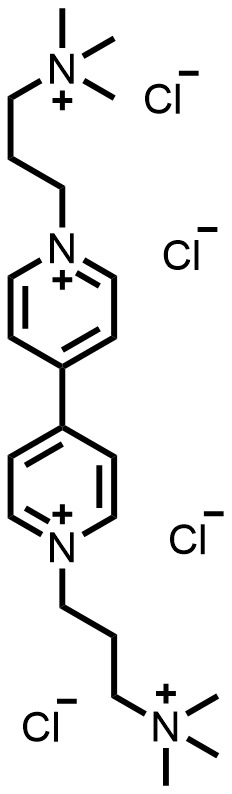 | 1.19 | 116 | 60 | 15.2 | NA/10.5 | NA/0.28 | [43] | |
| 2024 | 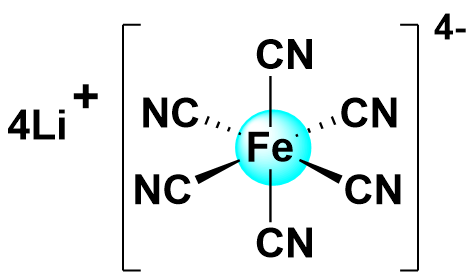 | Zn | 1.44 | NA | 86.09 | 61.64 | 400/24 | 0.0056/0.09 | [43] | |
“NA”, Not Available, 表示无相关数据报道. |
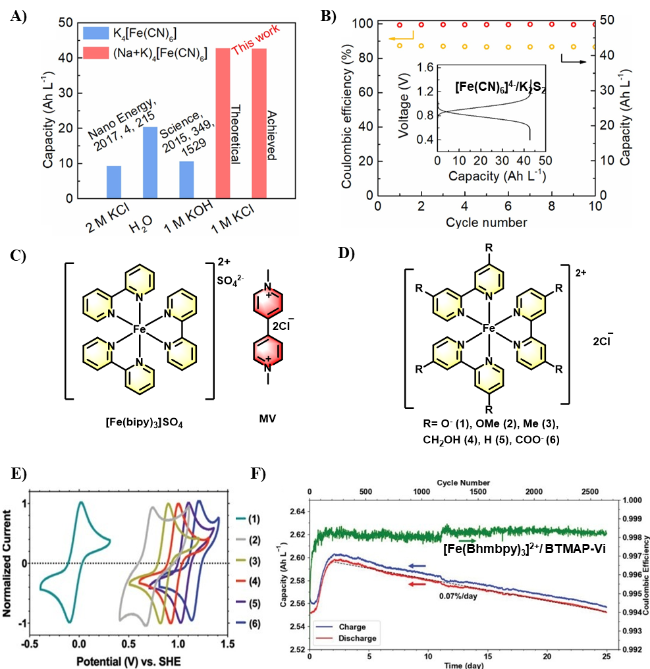
图5 A)不同离子[Fe(CN)6]4−浓度及其溶液电池容量密度的比较[40]; B) 1.6 mol/L [Fe(CN)6]4−/K2S2液流电池进行充放电循环[40]; C) [Fe(bipy)3]2+/MV结构式; D) [Fe(py)3]2+化学结构式; E) [Fe(py)3]2+的循环伏安图[42]; F) [Fe(Bhmbpy)3]2+/BTMAP-Vi液流电池循环测试[42]Figure 5 A) Comparison of different [Fe(CN)6]4− concentrations and their volumetric capacity[40]; B) Cycling performance of 1.6 mol/L [Fe(CN)6]4−/K2S2 AORFB[40]. Adapted with permission from [40] copyright 2021 Elsevier Science & Technology Journals. C) Chemical structure of [Fe(bipy)3]2+/MV; D) Chemical structure of [Fe(py)3]2+ derivatives; E) CV of [Fe(py)3]2+ derivatives[42]; F) Cycling performance of the [Fe(Bhmbpy)3]2+/BTMAP-Vi AORFB[42]. Adapted with permission from [42] copyright 2022 John Wiley and Sons |

图6 A)不对称铁配合物结构式; B)不同铁配合物的循环伏安图[41]; C) Na4[FeII(Dcbpy)2(CN)2]/(SPr)2V液流电池测试代表性的充电/放电容量电压图[41]; D) Na4[FeII(Dcbpy)2(CN)2]/(SPr)2V液流电池进行循环测试[41]; E) FeII(TPEN)Cl2的结构式Figure 6 A) Structural formulae of asymmetric iron complexes; B) Cyclic voltammograms of different iron complexes[41]; C) Representative voltage vs capacity traces of selected cycles for Na4[FeII(Dcbpy)2(CN)2]/(SPr)2V AORFB[41]; D) Cycling performance of the Na4[FeII(Dcbpy)2(CN)2]/(SPr)2V AORFB[41]. Adapted with permission from [41] copyright 2021 Springer Nature; E) Structural formulae of FeII(TPEN)Cl2 |
2.3 氮氧自由基类化合物

图7 A) TEMPO的降解过程图; B) TMAP-TEMPO/BTMAP-Vi液流电池示意图[22]; C) TMAP-TEMPO/BTMAP-Vi液流电池进行循环测试[22]Figure 7 A) Degradation reaction of TEMPO; B) Schematic representation of the TMAP-TEMPO/BTMAP-Vi AORFB[22]; C) Cycling performance of the TMAP-TEMPO/BTMAP-Vi AORFB[22]. Adapted with permission from [22] copyright 2019 Elsevier |
表3 TEMPO衍生物作为正极储能活性材料应用于水系有机液流电池Table 3 TEMPO derivatives used as posolyte in AORFBs |
| 年份 | 正极 | 负极 | 电压/V | 功率密度/ (mW•cm−2) | 能量效率/% | 容量密度/ (Ah•L−1) | 循环圈数/ 时间/d | 容量衰减率 每圈/每天/% | Ref. | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2015 |  | 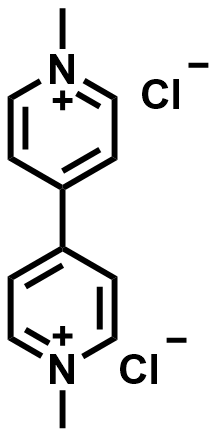 | 1.25 | NA | 61.5 | 56.3 | 100/NA | 0.11/NA | [21] | ||||||||||||||||||||
| 2015 | 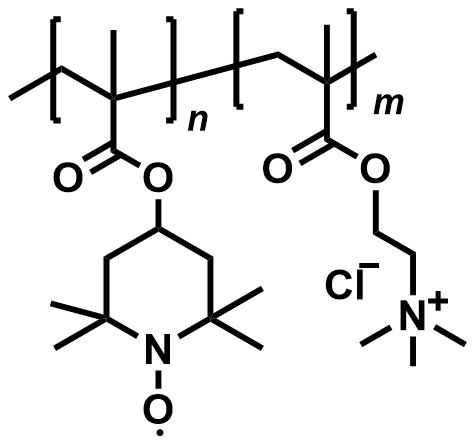 | 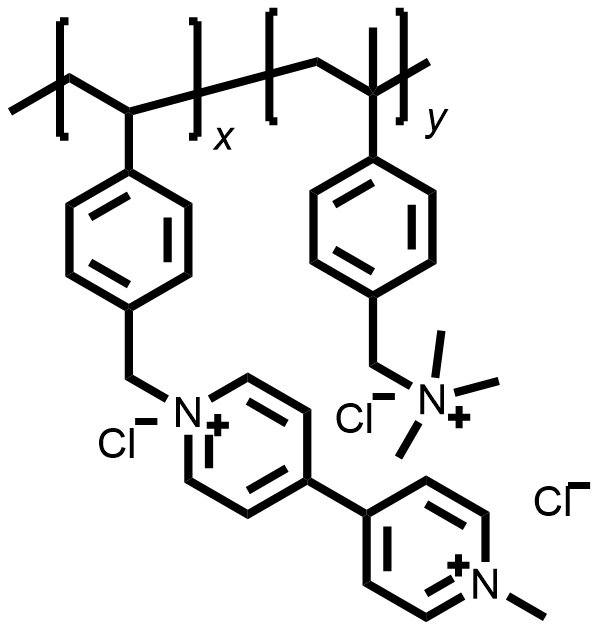 | 1.10 | NA | 80 | 10 | 10000/NA | NA/NA | [50] | ||||||||||||||||||||
| 2016 | 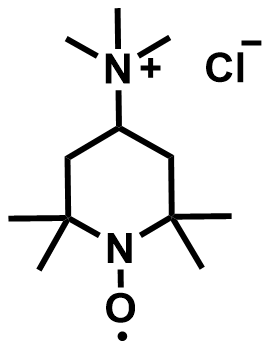 | 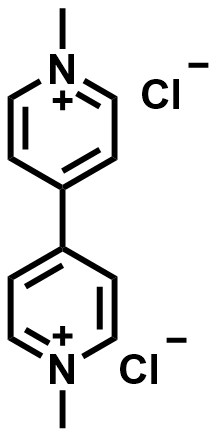 | 1.40 | NA | 75 | 54 | 100/NA | 0/NA | [51] | ||||||||||||||||||||
| 2017 | 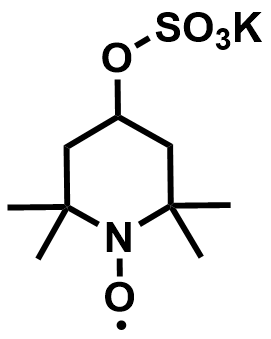 | Zn | 1.69 | NA | 85 | 26.7 | 1100/9 | NA/NA | [52] | ||||||||||||||||||||
| 2019 | 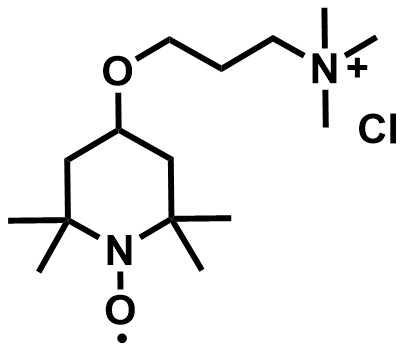 | 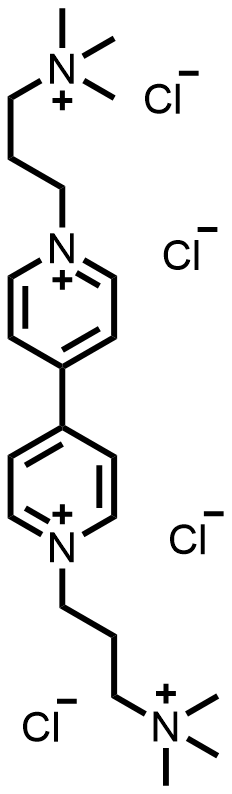 | 1.19 | 134 | 86 | 123.8 | 1000/NA | 0.007/NA | [22] | ||||||||||||||||||||
| 2019 |  | Zn | 1.64 | NA | 98.2 | NA | 400/NA | 0.55/NA | [53] | ||||||||||||||||||||
| 2021 | 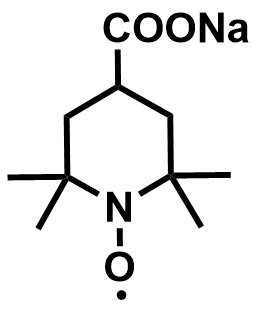 | 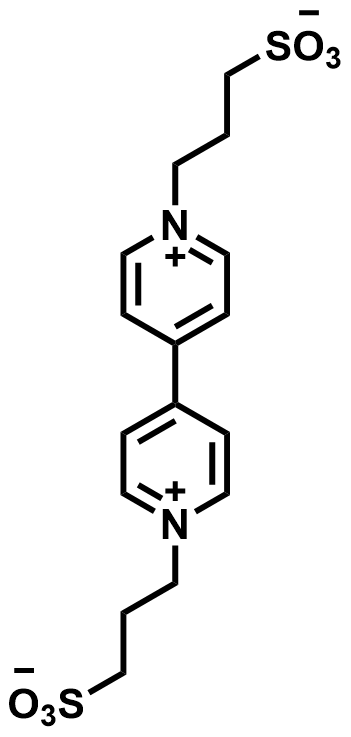 | 1.19 | NA | 64 | 67 | 400/NA | 0.06/NA | [54] | ||||||||||||||||||||
| 2021 | 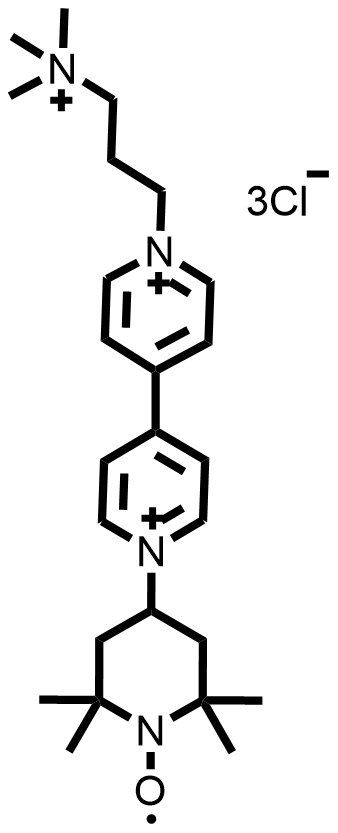 | 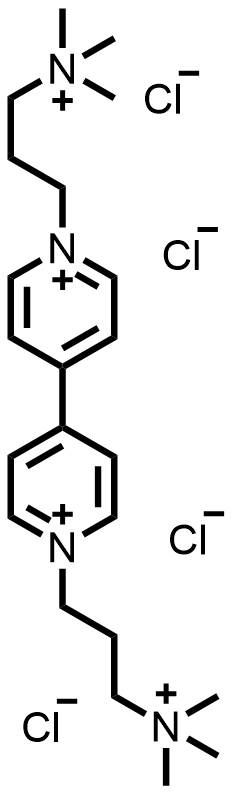 | 1.30 | NA | 70.6 | 37.6 | 100/NA | NA/NA | [55] | ||||||||||||||||||||
| 2021 | 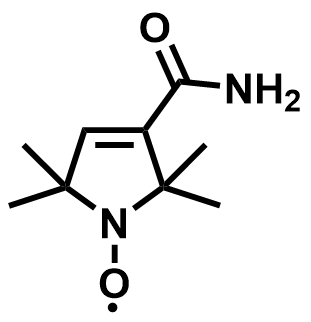 |  | 1.31 | NA | 78.5 | NA | 500/NA | 0.04/NA | [56] | ||||||||||||||||||||
| 2022 | 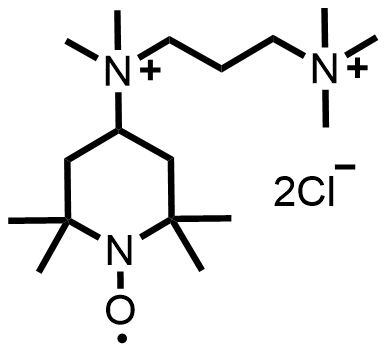 |  | 1.35 | 114 | 70.3 | 80.4 | 400/NA | 0/NA | [48] | ||||||||||||||||||||
| 2022 | 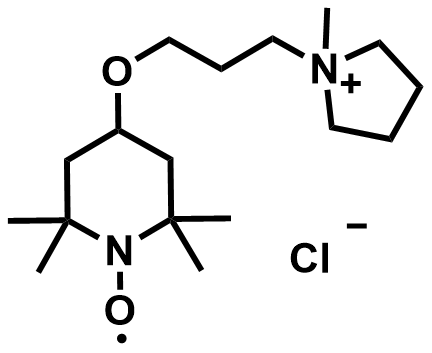 | 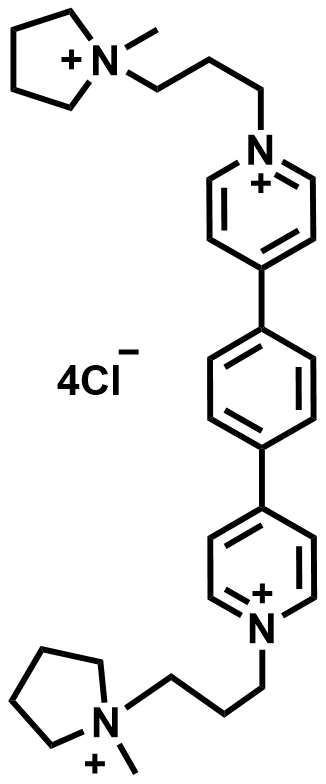 | 1.57 | 317 | 84 | 89.8 | 1000/14 | 0.05/3.47 | [57] | ||||||||||||||||||||
| 2022 | 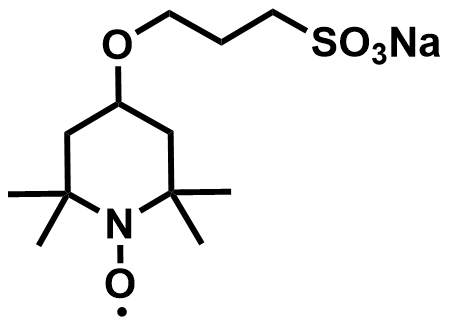 | 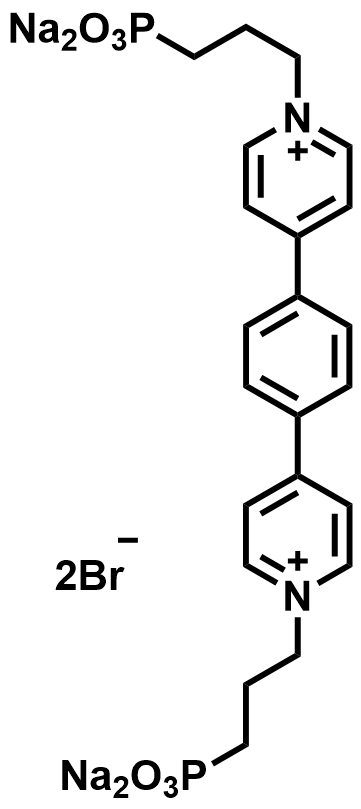 | 1.61 | 509 | 80 | 53.6 | 1200/16 | 0.021/0.7 | [58] | ||||||||||||||||||||
| 2022 | 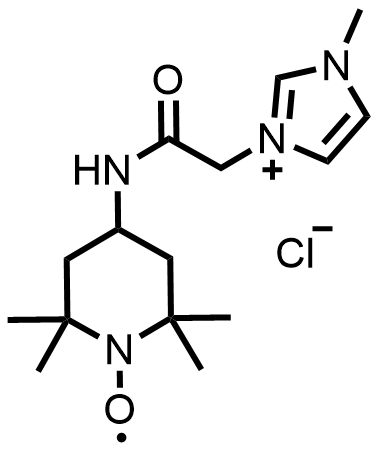 | Zn | 1.71 | 186.7 | 65.5 | 40.2 | 120/16.7 | 0.0069/0.05 | [59] | ||||||||||||||||||||
| 2023 | 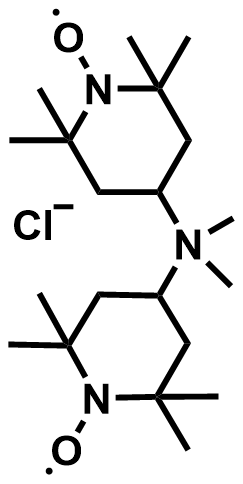 | 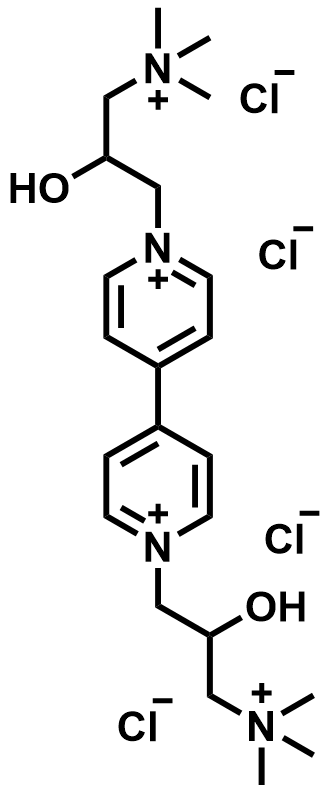 | 1.22 | 325 | 86.1 | 101 | 400/96 | 0/0 | [60] | ||||||||||||||||||||
| 2025 | 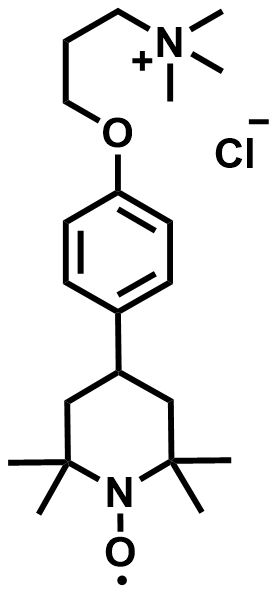 | 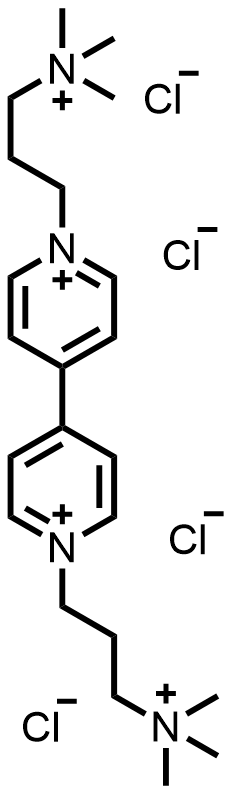 | 1.12 | 135 | 82 | 61.6 | 300/NA | 0.0018/NA | [61] | ||||||||||||||||||||
| 2025 | 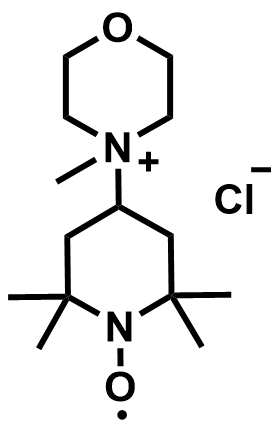 | 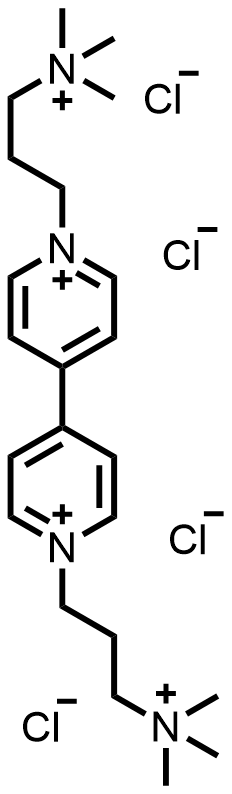 | 1.32 | 196 | 74.6 | 56 | 200/NA | 0.04/0.48 | [62] |
“NA”, Not Available, 表示无相关数据报道. |

图8 A) Pyr-TEMPO/[PyrPV]Cl4液流电池示意图[57]; B) Pyr-TEMPO/[PyrPV]Cl4循环伏安图[57]; C) (TPABPy)Cl3的循环伏安图[63]; D) MIAcNH-TEMPO的循环伏安图[59]; E) 4-CO2Na-TEMPO的循环伏安图[54]; F) 4-SO3K-TEMPO的循环伏安图[52]Figure 8 A) Schematic representation of the Pyr-TEMPO/[PyrPV]Cl4 AORFB[57]; B) CV of Pyr-TEMPO/[PyrPV]Cl4[57]. Adapted with permission from [57] copyright 2022 John Wiley and Sons; C) CV of (TPABPy)Cl3[63]. Adapted with permission from [63] copyright 2019 John Wiley and Sons; D) CV of MIAcNH-TEMPO[59]. Adapted with permission from [59] copyright 2022 John Wiley and Sons; E) CV of 4-CO2Na-TEMPO[54]. Adapted with permission from [54] copyright 2021 American Chemical Society; F) CV of 4-SO3K-TEMPO[52]. Adapted with permission from [52] copyright 2017 American Chemical Society |

图9 A) CPL, CPD和CT的循环伏安曲线图[56]; B) TMA-TEMPO和TPP-TEMPO的循环伏安曲线图[61]; C) TPP-TEMPO/BTMAP-Vi液流电池进行循环测试[61]; D) Mor-TEMPO和BTMAP-Vi的循环伏安曲线图[62]; E) Mor-TEMPO/BTMAP-Vi液流电池进行循环测试[62]Figure 9 A) CV curves of CPL, CPD and CT[56]. Adapted with permission from [56] copyright 2021 John Wiley and Sons; B) CV curves of TMA-TEMPO and TPP-TEMPO[61]; C) Cycling performance of the TPP-TEMPO/BTMAP-Vi AORFB[61]. Adapted with permission from [61] copyright 2025 Springer Nature; D) CV curves of Mor-TEMPO and BTMAP-Vi[62]; E) Cycling performance of the Mor-TEMPO/BTMAP-Vi AORFB[62]. Adapted with permission from [62] copyright 2025 Elsevier |
2.4 醌类化合物、吩噻嗪类化合物和其他自由基类化合物
表4 醌类化合物、吩噻嗪化合物和其他自由基类作为正极储能活性材料应用于水系有机液流电池Table 4 Quinone derivatives, phenothiazine derivatives, and other radical compounds used as posolyte in AORFBs |
| 年份 | 正极 | 负极 | 电压/V | 功率密度/ (mW•cm−2) | 能量效率/% | 容量密度/ (Ah•L−1) | 循环圈数/ 时间/d | 容量衰减率 每圈/每天/% | Ref. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2016 | 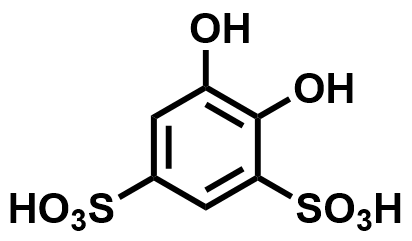 | 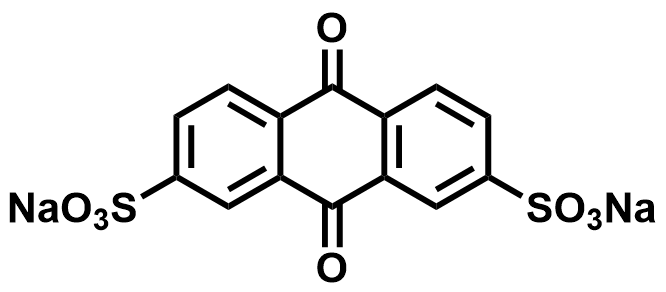 | 0.76 | NA | 70 | NA | 100/NA | NA | [64] | ||||||||||
| 2019 | 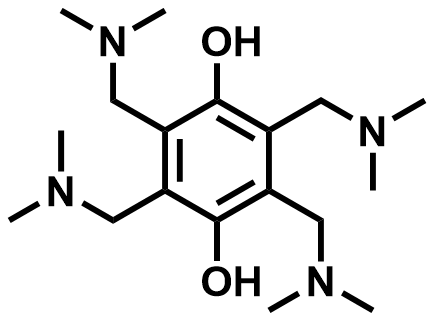 | Zn | 1.99 | NA | 70.2 | 5.36 | 50/NA | 0.08/2.22 | [65] | ||||||||||
| 2019 | 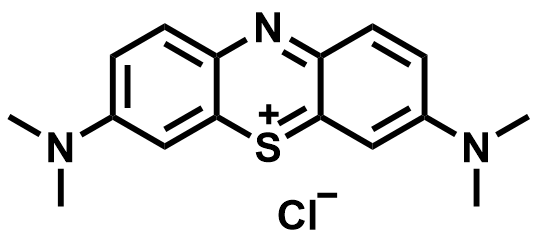 | V(II) | 1.12 | NA | 76 | 71 | 50/NA | 0.074/0.76 | [66] | ||||||||||
| 2021 | 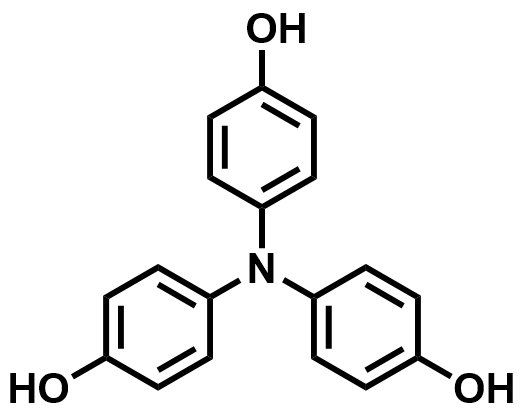 | Zn | 1.40 | 230 | 80 | 22.5 | 300/NA | NA/NA | [67] | ||||||||||
| 2022 | 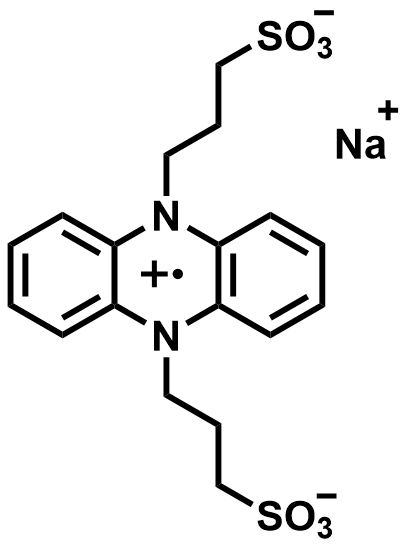 | Zn | 1.00 | 53 | 76.5 | 2.68 | 2500/27 | 0/0 | [68] | ||||||||||
| 2023 | 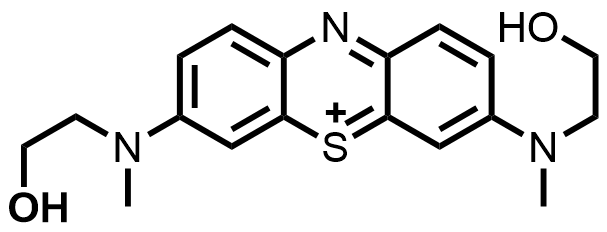 | V(II) | 1.10 | NA | 70 | 47 | 300/22 | 0.0056/0.077 | [69] | ||||||||||
| 2024 | 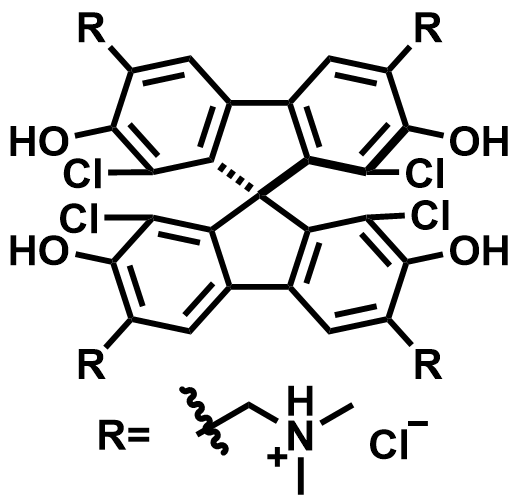 | H4[SiW12O40] | 1.05 | 230 | 83 | 37.5 | 250/22 | 0.05/0.56 | [70] | ||||||||||
| 2025 | 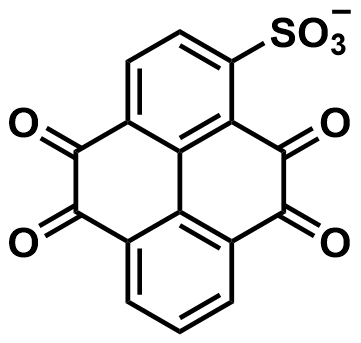 | V(II) | NA | 203.78 | 86 | 89 | 5200/60 | 0/0 | [71] | ||||||||||
| 2025 | 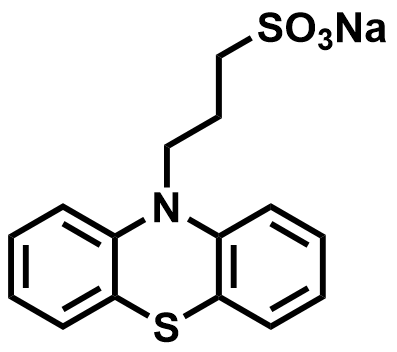 | 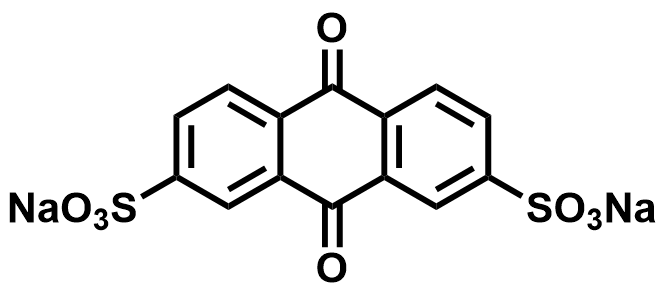 | 0.51 | NA | 72.55 | 52.26 | 1000/NA | NA/NA | [72] | ||||||||||
“NA”, Not Available, 表示无相关数据报道. |

图10 A)吩噻嗪衍生物循环伏安曲线图[66]; B)空气中稳定的MBH22+自由基; C) BHAP/V液流电池循环测试[69]Figure 10 A) CV curves of PTZ-based derivatives[66]. Adapted with permission from [66] copyright 2019 John Wiley and Sons; B) Air-stable MBH22+ radical; C) Cycling performance of the BHAP/V AORFB stacks[69]. Adapted with permission from [69] copyright 2023 John Wiley and Sons |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图11 A) THTPA双电子氧化还原过程[67]; B) PSPR的循环伏安图[68]; C) PSPR/Zn液流电池的循环测试[68]; D) SFA-Cl转化为CSFA-Cl的氯化反应过程[70]; E) PTO-PTS/V液流电池的循环测试[71]Figure 11 A) Two-electron transfer redox pathway of THTPA[67]. Adapted with permission from [67] copyright 2021 American Chemical Society; B) CV of PSPR[68]; C) Cycling performance of the PSPR/Zn AORFB[68]. Adapted with permission from [68] copyright 2022 American Chemical Society; D) Proposed reaction pathway from SFA-Cl to CSFA-Cl[70]. Adapted with permission from [70] copyright 2024 John Wiley and Sons; E) Cycling performance of the PTO-PTS/V AORFB[71]. Adapted with permission from [71] copyright 2025 American Chemical Society |