

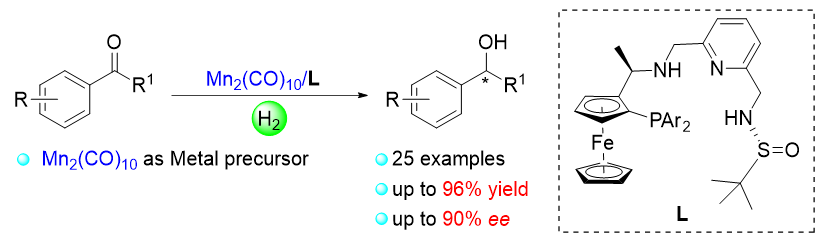
1 引言
手性醇类化合物是开发药物、农药、香精香料以及其他精细化学品的重要手性片段(图1, 左), 可以作为关键中间体衍生出各类精细化学品[1]. 因此, 开发一种经济、高效且环保的手性醇合成途径一直是学术界和工业界持续关注的研究热点. 过渡金属催化的前手性酮的不对称氢化(AH)反应是制备手性醇最有效的合成方法, 特别是以贵金属如铱(Ir)、钌(Ru)和铑(Rh)为催化剂的酮类AH反应[2], 如Noyori等[3]开发的金属钌催化剂、周其林课题组[2a-2c]与张绪穆课题组[2g,2h,4]基于金属铱开发的多齿配体催化体系, 在酮的AH反应中都体现了超高的催化活性和对映选择性. 然而, 贵金属催化剂的高成本和有限的资源储量限制了其大规模工业应用. 此外, 贵金属催化剂存在金属残留问题, 这对医药和食品领域的产品质量是非常严峻的挑战. 进入21世纪以来, 随着绿色化学和可持续发展理念深入人心, 廉价金属(如铁、钴、镍和锰等)催化体系[5]受到了越来越多的关注. 廉价金属不仅储量丰富、价格低廉, 而且环境友好, 具有替代贵金属催化剂的潜力.

近年来, 锰催化前手性酮的AH合成手性醇[6]的研究取得了显著进展. 这一领域的快速发展得益于2017年Clarke课题组[7]和Beller课题组[8]采用手性三齿配体与Mn(CO)5Br生成的锰催化剂, 实现了锰催化酮不对称氢化的突破性工作. 紧接着, 我国科学家相继报道了多例锰催化酮的AH反应, 大大推动了这一领域的发展. 如2019年, 丁和韩等[9]开发了一系列高度模块化的手性钳形配体与Mn(CO)5Br形成催化剂用于酮类化合物的不对称氢化. 该配合物展现了前所未有的高活性[可达9800 TON (转化数, turnover number)]、广泛的底物范围(81例)、良好的官能团耐受性和优异的对映选择性(85%~98% ee). 2020年, 张课题组[10]合成了一类二茂铁类PNP三齿配体与Mn(CO)5Br络合形成一个七元环锰催化剂, 在简单酮的AH中获得了优异的催化活性(2000 TON)和对映选择性(高达99% ee). 钟及其团队发展了基于二茂铁的PNN三齿配体与Mn(CO)5Br的配合物在一系列的前手性酮类化合物的不对称氢化工作中也取得了突出的成绩, 成功实现了锰催化简单酮[11](对映选择性高达88.5% ee, TON高达8200)、α,β-不饱和酮[12](对映选择性66%~86% ee, TON 9500)以及β-酮砜[13](对映选择性高达97% ee)的AH反应, 此外, 该类催化剂也能以高对映选择性(高达99% ee)和优异的催化活性(TON高达13000)实现前手性二苯甲酮[14]的不对称氢化. 王课题组[15]在2023年报道了一类二茂铁PNN三齿配体与Mn(CO)5Br生成手性催化剂, 在芳基酮的不对称氢化反应中取得了较好的成绩(对映选择性可达93% ee).
尽管锰催化简单酮的不对称氢化反应取得了一些重要进展, 但通过现有文献的调研发现, 已报道的锰催化酮的不对称氢化工作均采用五羰基溴化锰(Mn (CO)5Br)作为锰金属前体, 而目前商品化的羰基锰化合物有十羰基二锰(Mn2(CO)10)和五羰基溴化锰(Mn(CO)5Br), 从价格与合成难度的角度来分析, 十羰基二锰(Mn2(CO)10)是用于不对称催化反应的更优选择, 可以进一步降低用于不对称氢化反应的金属催化剂成本. 而且合成Mn(CO)5Br时[16]需要用到CCl4和Br2, 所以使用Mn2(CO)10作为金属前体也能够减少有毒有害试剂的使用, 避免有害物质排放, 是更加绿色环保的不对称催化氢化反应. 基于此, 本课题以二茂铁为骨架, 引入手性叔丁基亚磺酰胺[17], 设计并合成了一系列手性PNNN四齿配体, 与Mn2(CO)10原位络合生成低价态锰(Mn(0))催化剂, 实现了以Mn2(CO)10为金属前体的锰催化简单酮的不对称氢化反应(图1, 右).
2 结果与讨论
2.1 新型二茂铁配体的制备
配体合成路线反应式如图2. 从商业化的Ugi’s胺(1)出发, 按照课题组之前报道的方法[18]经两步反应快速合成(R,S)-3. (R,S)-3与化合物(S)-5[19]在甲醇中发生还原胺化反应快速制得目标配体(R,S,S)-6a~(R,S,S)-6f. 我们采用(R)-叔丁基亚磺酰胺同时制备了配体(R,S,R)-6g用于研究此类配体的构型匹配效应. 以苯乙酮为标准底物, nsubstrate(S)/nbase(B)=10/1的叔丁醇钾(KOtBu)为碱, 选择配体6a和6g分别和金属[Ir(COD)Cl]2络合成催化剂[nS/nC=50/1, (catalyst, C)]在MeOH中进行不对称氢化反应(表1, Entries 1, 2), 实验结果显示, 配体6a无论是转化率还是对映选择性都远高于配体6g, 说明配体(R,S,S)-6a的构型是匹配构型. 本文之后用于不对称催化氢化反应的配体构型均为(R,S,S).
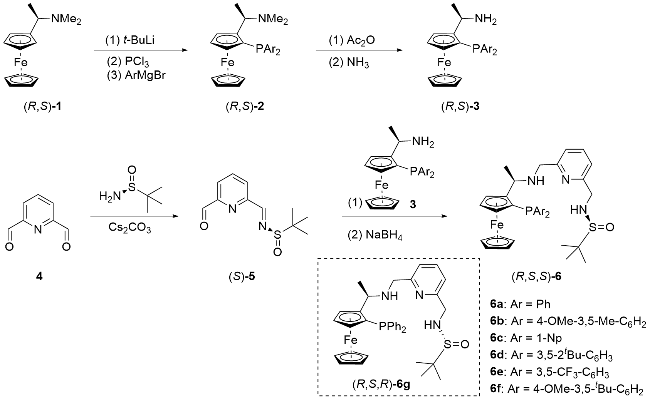
表1 不对称氢化反应的金属和配体筛选Table 1 Metal precursor and ligand screening for asymmetric hydrogenation reactions |

| Entry | 配体 | 金属前体 | 转化率a/% | 对映选择性eeb/% |
|---|---|---|---|---|
| 1 | 6a | [Ir(COD)Cl]2 | 99 | 37 |
| 2 | 6g | [Ir(COD)Cl]2 | 65 | 14 |
| 3 | 6a | Mn(CO)5Br | 99 | 69 |
| 4 | 6a | Mn2(CO)10 | 99 | 67 |
| 5 | 6b | Mn2(CO)10 | 99 | 59 |
| 6 | 6c | Mn2(CO)10 | — | — |
| 7 | 6d | Mn2(CO)10 | 99 | 77 |
| 8 | 6e | Mn2(CO)10 | 47 | 79 |
| 9 | 6f | Mn2(CO)10 | 99 | 67 |
| 10 | 6g | Mn2(CO)10 | 99 | 56 |
a The conversion rate was determined by 1H NMR. b The enantioselectivity was determined by HPLC. |
在合成得到PNNN四齿配体6a~6f后, 我们对这些配体在锰催化酮不对称氢化中的催化效果进行了初步尝试, 以苯乙酮为标准底物, 在叔丁醇钾作碱, 甲醇为溶剂的条件下, 以nS/nC=50/1的催化剂负载量, 在5 MPa H2压力、50 ℃下进行了不对称氢化反应, 结果如表1所示, 从Entries 1~4可得出, 在苯乙酮的AH中, 配体6a与金属锰的配合物展现出比与金属铱配合物更高的对映选择性, 产物ee值可以达到69% ee和67% ee. 值得注意的是, 此类配体可以与Mn2(CO)10形成配合物有效地催化苯乙酮的不对称氢化, 紧接着, 在室温下以Mn2(CO)10为金属前体分别与配体6b~6f原位络合成锰配合物在相同条件下测试了一系列配体的催化性能(Entries 5~9), 除配体6c外, 其它配体都能将苯乙酮不对称转化为手性苯乙醇, 配体6d、6e具有较大位阻基团, 对映选择性最高, 分别为77% ee 和79% ee, 但配体6e催化的反应转化率较低, 只能达到47%. 综合起来, 配体6d在以Mn2(CO)10为前体的锰催化苯乙酮的不对称氢化反应中的催化性能最好, 转化率可以达到99%以上, 产物ee值也能达到77%.
2.2 不对称氢化反应条件的优化
氢化反应条件如溶剂、碱、温度和H2压力等对催化剂的立体选择性有着重要的影响. 配体筛选实验结果表明配体6d在苯乙酮的不对称氢化中表现最优, 因此我们以6d为手性配体, 以Mn2(CO)10为锰金属前体, 苯乙酮为标准底物对氢化反应条件进行了优化(表2). 实验结果显示, 极性醇类溶剂是这个反应的优良溶剂, 以甲醇为溶剂时立体选择性较乙醇低, 在非极性的醚类溶剂做反应溶剂时未发现手性产物. 随后, 以乙醇为溶剂在同等反应条件下, 考察了不同的碱对氢化反应的影响(Entries 8~16), 如表2所示, 由于Na2CO3、K2CO3和Cs2CO3三种碱在乙醇中溶解性较差, 这三种碱参与的氢化反应转化率较差, 都没能实现苯乙酮完全转化为苯乙醇, 同样LiOMe参与的氢化反应转化率也只有78%. 以KOtBu, LiOtBu, NaOtBu和NaOH作碱的氢化反应均使得苯乙酮全部转化为苯乙醇, 并取得了较高的对映选择性, 分别达到了77% ee, 78% ee, 76% ee和78% ee. 当将催化剂负载量降到nS/nC=100/1, 且反应温度和氢气压力进一步降低时(Entry 17), 苯乙酮也能以78% ee的对映选择性完全转化为手性苯乙醇. 当在以叔丁醇钾为碱的氢化反应中添加六氟异丙醇(nS/nHFIP=10/1)后(Entry 18), 反应立体选择性提升到了84% ee. 最终确定了最佳的反应条件为: 以配体6d和Mn2(CO)10 (nS/nC=100/1) 在乙醇中原位络合生成手性催化剂, 在氢气压力3.5 MPa, 反应温度35 ℃下, 以EtOH为溶剂, KOtBu (nS/nB=10/1, 添加六氟异丙醇(nS/nHFIP=10/1)后反应24 h, 此催化体系能够发挥出最好的催化性能.
表2 不对称氢化反应条件优化Table 2 Optimization of conditions for asymmetric hydrogenation |
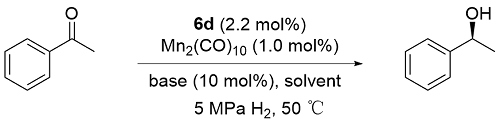
| Entry | 溶剂 | 碱 | 转化率a/% | 对映选择性eeb/% |
|---|---|---|---|---|
| 1 | MeOH | KOtBu | 99 | 75 |
| 2 | iPrOH | KOtBu | 58 | 70 |
| 3 | EtOH | KOtBu | 99 | 77 |
| 4 | TFE | KOtBu | 18 | 68 |
| 5 | THF | KOtBu | NR | — |
| 6 | HFIP | KOtBu | NR | — |
| 7 | MTBE | KOtBu | NR | — |
| 8 | EtOH | NaOtBu | 99 | 76 |
| 9 | EtOH | LiOtBu | 99 | 78 |
| 10 | EtOH | Na2CO3 | 58 | 83 |
| 11 | EtOH | K2CO3 | 62 | 81 |
| 12 | EtOH | Cs2CO3 | 95 | 77 |
| 13 | EtOH | NaOMe | 98 | 77 |
| 14 | EtOH | LiOMe | 78 | 78 |
| 15 | EtOH | NaOH | 98 | 78 |
| 16 | EtOH | KOH | 99 | 78 |
| 17c | EtOH | KOtBu | 99 | 78 |
| 18d | EtOH | KOtBu | 99 | 84 |
a The conversion rate was determined by 1H NMR. b The enantioselectivity was determined by HPLC. c nS/nC=100/1, 3.5 MPa H2, 35 ℃. d nS/nC=100/1, 3.5 MPa H2, 35 ℃, 10 mol% HFIP. |
2.3 底物普适性研究

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
从取代基的电性角度去分析, 对苯环上被吸电子基团取代的底物(8-6~8-11)加氢的对映选择性要低于被给电子基团取代的底物(8-4~8-5, 8-12~8-14), 且吸电子能力越强, 对映选择性越低, 如强吸电子基团取代的底物(8-10)加氢反应的对映选择性只有71% ee, 而强给电子基团取代底物(8-14)的加氢反应对映选择性达到了90% ee. 此外, 对R1位置不同取代基对加氢反应的影响也进行了考察, 当该位置取代基团越大, 加氢反应的对映选择性相对越低, 如取代基为乙基时, 加氢产物8-15的对映体过量值可以达到89% ee, 但当取代基为位阻较大的叔丁基时, 加氢产物8-18的对映体过量值只有73% ee. 此催化体系也能顺利完成苯并环酮的不对称氢化(8-21, 8-22和8-25), 并以较高的对映选择性(74%~86% ee)获得手性产物. 此催化体系还能以较高的对映选择性对萘乙酮进行不对称加氢反应, 产物8-23与8-24对映体过量值分别为83% ee和85% ee. 当R1也是芳基时(8-20), 由于酮两侧基团相似, 所以此类底物的不对称氢化较难, 但此催化体系依然能够以高产率和较高的对映选择性(82% ee)对此类底物完成不对称氢化.
3 结论
综上, 本工作设计并合成了一类新型手性二茂铁骨架PNNN四齿配体, 并与Mn2(CO)10在室温下原位络合制备成零价锰催化剂, 该催化剂在一系列不同位阻/电性效应基团取代芳香酮的不对称氢化反应中能够获得较高的产率和良好的对映选择性(45%~90% ee). 虽然该催化体系在活性和选择性方面仍有较大的进步空间, 但是本工作实现了以Mn2(CO)10为锰前体催化酮的不对称氢化反应, 提示我们可以通过进一步优化配体结构, 改善配体与Mn2(CO)10的配位环境以提高锰催化不对称氢化反应的活性和对映选择性, 发展出更加经济、环保、高效的手性锰催化剂.
(Lu, Y.)