


1 引言
2 结果与讨论
2.1 金纳米形貌对反应的影响
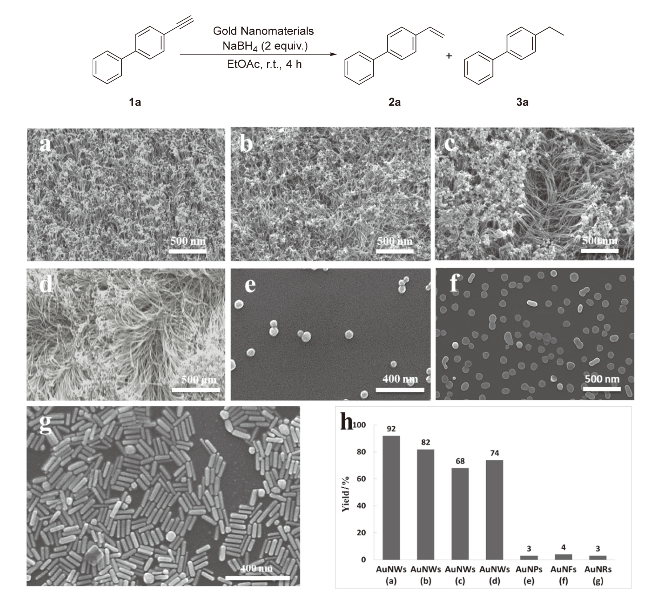
图1 使用1,3,5-三乙炔基苯配体制备的长度为(a) 452±12 nm、(b) 1338±34 nm和(c) 2446±63 nm的AuNWs的扫描电子显微镜(SEM)图; (d)使用3-巯基苯甲酸(3-MBA)配体制备的长度为1000±23 nm的AuNWs的SEM图; (e) 60 nm 的AuNPs的SEM图; (f)长度为100 nm的AuNFs的SEM图; (g)长度为115 nm的AuNRs的SEM图; (h) 金纳米材料的催化效率(催化剂用量约为0.76 mol%), 总产率为烯烃(2a)和烷烃(3a)的HPLC产率之和(测量3次取平均值)Figure 1 SEM images of AuNWs with (a) 452±12 nm length, (b) 1338±34 nm length, and (c) 2446±63 nm length using 1,3,5-triethynylbenzene ligand, and (d) 1000±23 nm length using 3-mercaptobenzoic acid (3-MBA) ligand; SEM images of (e) 60 nm AuNPs, (f) 100 nm (length) AuNFs, and (g) 115 nm (length) AuNRs; (h) catalytic efficiency of gold nanomaterials (catalyst loading of 0.76 mol%). The yield is overall HPLC yield of alkenes (2a) and alkanes (3a) (average value of 3 times) |
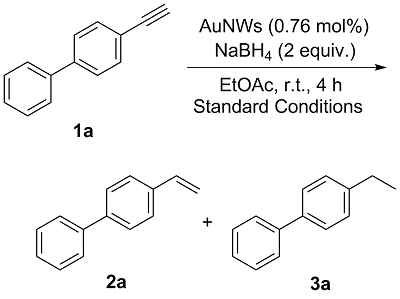
2.2 反应条件的优化
表1 反应条件的优化aTable 1 Optimization of reaction conditionsa |
| Entry | 与标准条件的差异 | 产率b/% | 2a与3a的 物质的量比b |
|---|---|---|---|
| 1 | 无 | 92 | 72/28 |
| 2 | NaBH4 (1 equiv.) | 88 | 84/16 |
| 3 | NaBH4 (3 equiv.) | 90 | 80/20 |
| 4 | 无AuNWs | 无反应 | — |
| 5 | AuNWs (0.38 mol%) | 90 | 88/12 |
| 6 | AuNWs (1.52 mol%) | 89 | 74/26 |
| 7 | AuNWs (2.28 mol%) | 49 | 95/5 |
| 8 | EtOH替代EtOAc | 9 | — |
| 9 | DMSO替代EtOAc | 6 | — |
| 10 | CH2Cl2替代EtOAc | 微量 | — |
| 11 | THF替代EtOAc | 65 | 65/35 |
| 12 | H2替代NaBH4 | 无反应 | — |
a Reaction conditions: 4-ethynyl-1,1'-biphenyl (17.8 mg, 0.1 mmol), NaBH4 (7.56 mg, 2 equiv.) and AuNWs with 452±12 nm length (0.15 mg, 0.76 mol%) were mixed in EtOAc (1.5 mL) and stirred at room temperature for 4 h. b By HPLC analysis. |
2.3 反应随时间的演变趋势
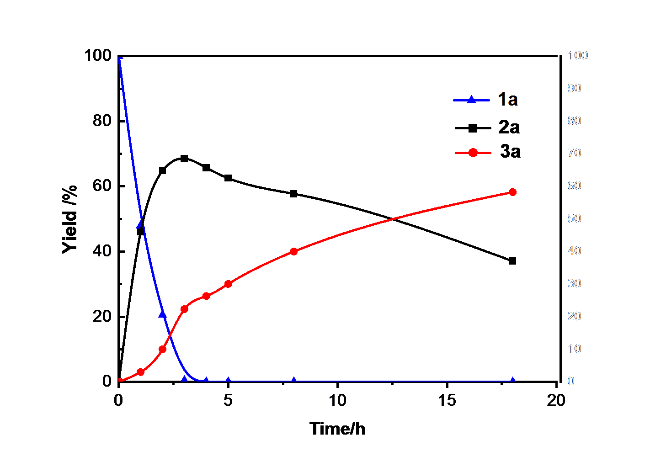
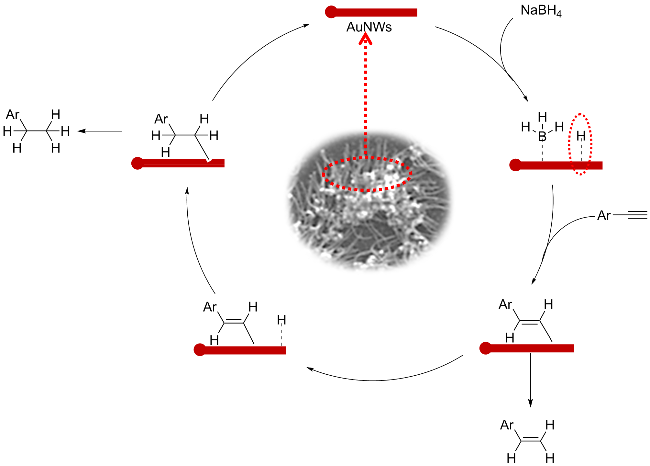
2.4 反应适用底物的探究
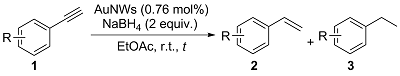
表2 碱促进的炔酰胺1的螺环化反应底物范围研究aTable 2 Substrate scope study for the base-promoted spirocyclization of ynamides 1a |
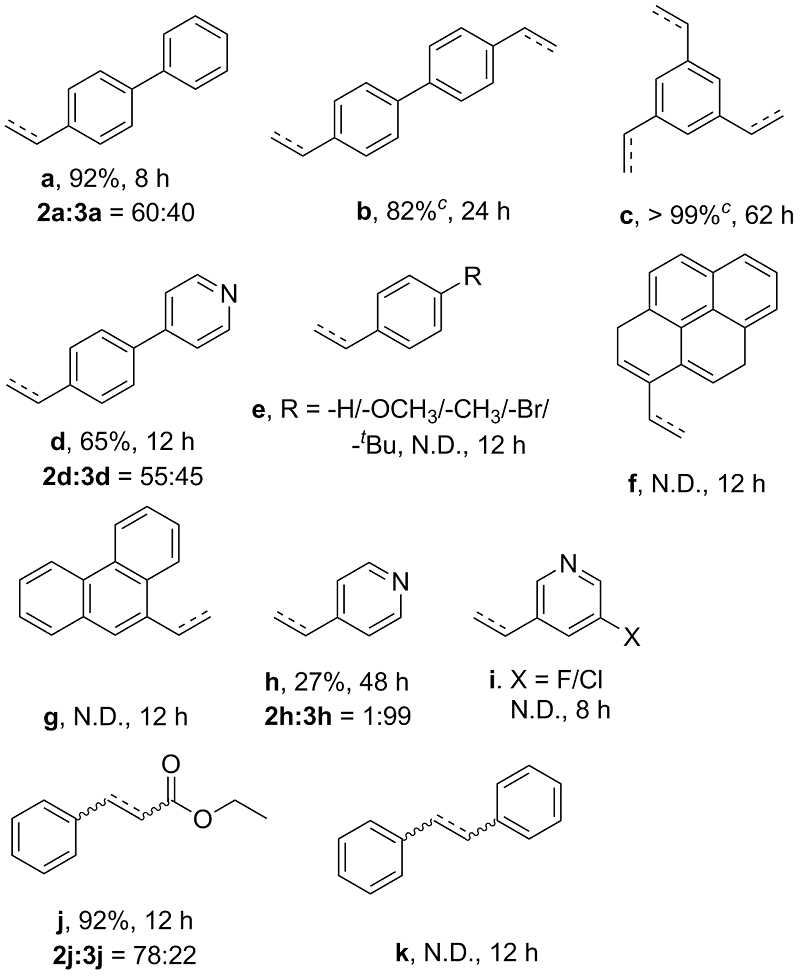 |
a Reaction conditions: aryl alkynes (0.1 mmol), AuNWs with 452±12 nm length (0.15 mg, 0.76 mol%) and NaBH4 (7.56 mg, 2 equiv.) were mixed in EtOAc (1.5 mL) and stirred at room temperature. b Overall yields and product ratios were determined by HPLC analysis. c A mixture of various hydrogenation products. N.D.=not detected. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}