

1 引言
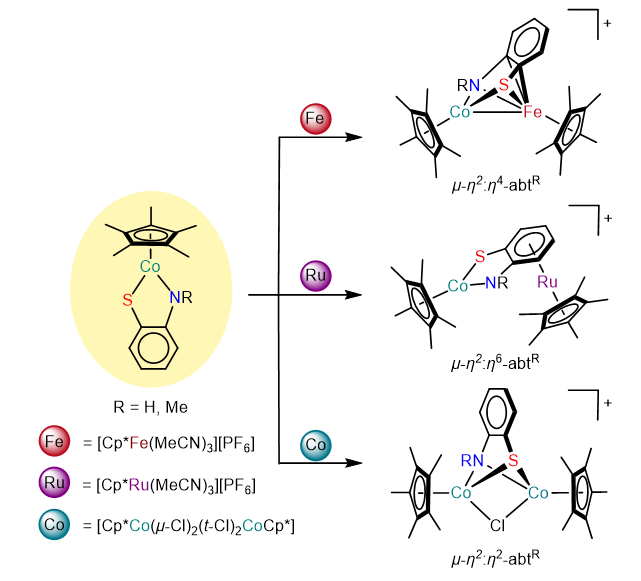
邻氨基苯硫酚(abt)及其衍生物(abtR)是氧化还原非保守性配体, 能够参与到氧化还原反应的电子传递过程中, 从而影响金属配合物的反应性能[3]. 同时, 配体的氨基可以质子化、去质子化以及烷基化, 还可通过与金属的动态配位调控配合物的反应性能[4]. 近十年来, 人们合成了一系列结构明确的邻氨基苯硫酚单核配合物并广泛应用于各个领域, 如电催化制氢[5]、构筑加氧酶模型[6]、制备磁性材料[7]等. 然而, abtR配体丰富的氧化还原活性使得双金属配合物, 尤其是异双核金属配合物的定向合成, 相较于单金属配合物更具挑战性. 如图1A所示, 2012年, Ruiz等[8]报道了首例邻氨基苯硫酚桥联双钼配合物[CpMo(t-CO)(μ-η1:η2-abt)(μ-PPh2)2MoCp] (I, Cp=η5-C5H5), 并研究了其在碱性溶液中去质子化反应. 目前报道的abtR桥联双核配合物仅有几例, 且多为配位饱和的二聚体(如II和III)[9], 既缺乏小分子配位活化的位点, 又存在稳定性差易分解为单核配合物的问题. 值得注意的是, 目前已报道的体系中, abtR普遍以μ-η1:η2形式桥联双金属中心, 仅有一例其他配位形式的abtR桥联双核配合物的报道[8].
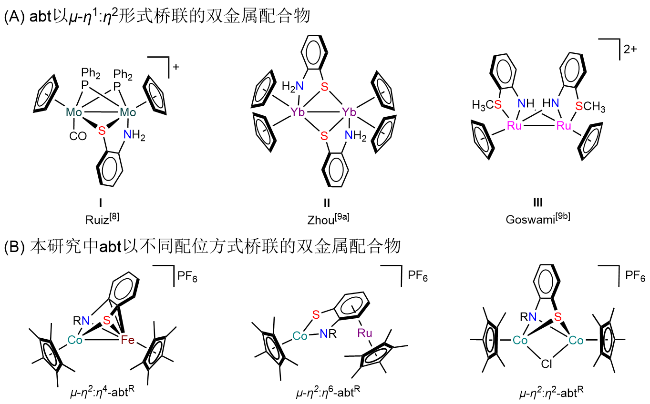
在本课题组前期工作中, 设计合成了一系列新型硫桥联同/异双核金属配合物, 利用金属间协同作用, 成功实现了多种小分子的活化和转化[10-14]. 其中, 以邻苯二硫酚(bdt)为配体, 设计合成的一系列新型双金属配合物展示出了优异的反应性能, 如bdt桥联双铁配合物[Cp*Fe(μ-η2:η4-bdt)FeCp*] (Cp*=η5-C5Me5)作为新型固氮酶模拟物, 实现了由二氮烯逐步还原产氨的功能模拟[12]. 而双钴配合物[Cp*Co(μ-η2:η2-bdt)(μ-I)CoCp*]- [PF6] (IV)则可以作为质子还原产氢的电催化剂[15]. 与bdt结构相似的abt配体因引入可质子化的氨基, 在催化反应中可能表现出更优的反应性能. 因此, 本文报道邻氨基苯硫酚桥联钴铁、钴钌和双钴配合物的合成、表征和反应性. 单晶X-射线衍射结果显示, abtR以三种不同的方式配位于双金属中心(图1B), 值得注意的是, 双钴配合物可以与叠氮钠反应, 生成结构稳定的双钴叠氮配合物. 进一步研究发现, 配体桥联方式的差异导致三种双金属配合物表现出截然不同的反应性.
2 结果与讨论
2.1 邻氨基苯硫酚桥联双核配合物的合成与表征
首先采用盐交换策略[10], 以[Cp*Co(μ-Cl)2CoCp*][16] 为前体, 与邻氨基苯硫酚锂盐反应. 产物为单核钴配合物[Cp*Co(abt)] (1a)[17], 并未得到硫桥联双核钴配合物. 这可能是由于CoIICoII配合物不稳定, 发生分解. 基于以下两点考虑: (1)本课题组前期通过组装策略成功合成了一系列硫桥联双金属配合物[18], 配合物1a的硫/氮原子存在孤对电子, 具有参与组装反应的潜力; (2)乙腈的配位能力较弱, 为反应保留了潜在配位点, 使反应在室温条件下即可进行[19], 从而有效规避高温生成二聚体副产物[9b]. 因此, 我们改变合成方法, 采用如图2的单核前体定向组装策略, 成功构筑了双金属配合物2[PF6]~4[PF6]. 采用与1a相似的合成方法, 通过[Cp*CoI2(CO)][20]与三乙胺、邻甲氨基苯硫酚(abtMe)反应, 得到了单核钴配合物[Cp*Co(abtMe)] (1b).
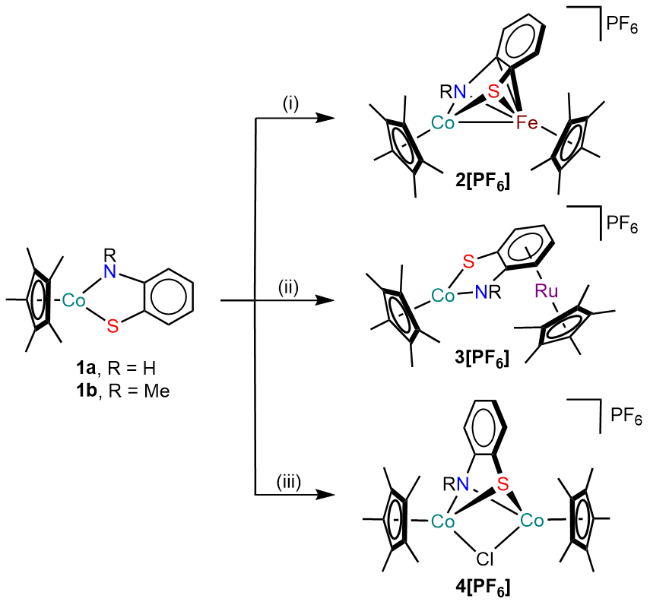
图2 配合物2[PF6]、3[PF6]和4[PF6]的合成aFigure 2 Synthesis of complexes 2[PF6], 3[PF6] and 4[PF6]a a Reaction conditions: (i) 1 equiv. [Cp*Fe(MeCN)3][PF6], CH2Cl2, -78 ℃ to r.t., 91% for 2a[PF6] and 92% for 2b[PF6]; (ii) 1 equiv. [Cp*Ru(MeCN)3]- [PF6], CH2Cl2, -78 ℃ to r.t., 98% for 3a[PF6] and 95% for 3b[PF6]; (iii) 1 equiv. [Cp*Co(μ-Cl)2(t-Cl)2CoCp*], 1 equiv. KPF6, MeOH, -78 ℃ to r.t., 82% for 4a[PF6] and 94% for 4b[PF6] |
2.1.1 异核配合物的合成与表征
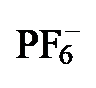
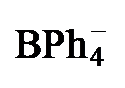
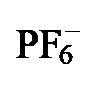
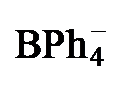
为进一步探究中心金属对配位模式的影响, 将金属前体调变为三乙腈钌[Cp*Ru(MeCN)3][PF6], 相同条件下分别以98%和95%收率得到了茂基钌结构单元与邻氨基苯硫酚苯环π配位的钴钌配合物[Cp*Co(μ-η2:η6- abtR)RuCp*][PF6] (3a[PF6], R=H; 3b[PF6], R=Me), 并未得到与本组之前报道的[Cp*Ru(μ-η2:η4-bdt)RuCp*]结构相似的钴钌配合物[21]. 这可能因为N原子的路易斯碱性强于S原子, 更有利于稳定16电子三明治型配合物, 导致邻氨基苯硫酚和邻苯二硫酚的配位方式存在差异[22]. 由于苯环和钌中心的π键作用, 一方面钌配位部分破坏了苯环的共轭结构, 环电流强度减弱, 去屏蔽效应降低; 另一方面, 金属作为电子供体, 增加了苯环的电子密度, 增强了氢周围的屏蔽效应, 最终导致苯环氢出峰位置向高场偏移到δ 4.92~5.95区间.
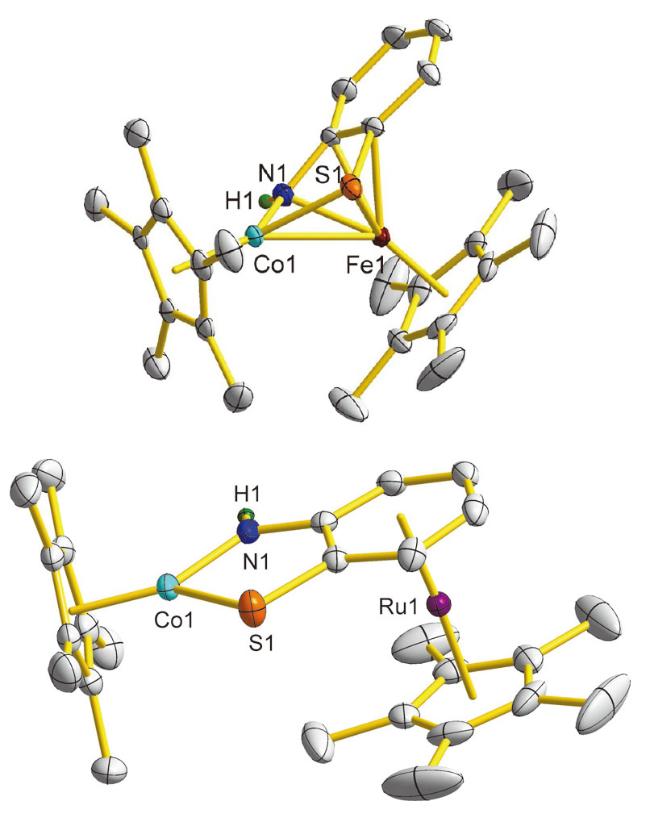
对以上两种异双核金属配合物的分子结构分析表明: 配合物2a[PF6]和2b[PF6]的Co…Fe间距(0.25925(9)和0.26156(6) nm)显著短于配合物V的Co…Fe间距(0.2796(1) nm)[15], 这预示着2[PF6]中钴、铁之间可能存在更强的金属/金属间作用. 邻氨基苯硫酚向一侧的铁中心倾斜导致2a[PF6]和2b[PF6]中两个Cp*所在平面形成的二面角分别为59.9(3)°和56.8(1)°(表1); 而在配合物3中, 钌配位的茂环与苯环近似平行(图3), 使3a[BPh4]和3b[BPh4]中两个Cp*所在平面几乎垂直, 二者所在平面的二面角分别为87.6(2)°和87.8(6)°. 这些关键的结构数据差异可能预示着两种配合物对相同的底物具有不同的反应性.
表1 配合物2[PF6]和3[BPh4]的主要结构数据Table 1 Main structural data of complexes 2[PF6] and 3[BPh4] |
| 2a[PF6] | 2b[PF6] | 3a[BPh4] | 3b[BPh4] | |
|---|---|---|---|---|
| 距离/nm | ||||
| Co…Fe | 0.25925(9) | 0.26156(6) | — | — |
| Co—N | 0.1872(5) | 0.1903(2) | 0.1858(3) | 0.1757(8) |
| Co—S | 0.2157(2) | 0.21589(8) | 0.2157(1) | 0.2191(3) |
| Co—Cp* | 0.16693(7) | 0.16807(5) | 0.16485(4) | 0.16763(7) |
| Fe/Ru—Cp* | 0.16869(7) | 0.16921(6) | 0.18099(4) | 0.18011(5) |
| 二面角/(°) | ||||
| Cp*1∠Cp*2 | 59.9(3) | 56.8(1) | 87.6(2) | 87.8(6) |
2.1.2 同核配合物的合成与表征
鉴于同核配合物可能具有不同的反应性能以及bdt桥联双钴配合物的催化活性[14-15], 进一步开展了邻氨基苯硫酚桥联双钴配合物的设计合成. 首先分别以[Cp*CoI2(CO)]和[Cp*Co(μ-Cl)2CoCp*]为前体与1进行反应, 采用与前期工作类似的合成方法未能高选择性获得双钴配合物[15]. 因此将反应前体调变为二聚体配合物[Cp*Co(μ-Cl)2(t-Cl)2CoCp*], 分别以82%和94%收率得到了双钴配合物[Cp*Co(μ-η2:η2-abtR)(μ-Cl)- CoCp*][PF6] (4a[PF6], R=H; 4b[PF6], R=Me)(图4). 配合物4存在一个桥联的氯离子, 具有潜在的反应活性位点, 可能具有活化小分子的能力[23]. 配合物4a[PF6]和4b[PF6]的氢核磁谱图中, 化学位移值在1.15和0.94的尖峰归属为Cp*上甲基的信号峰, 这表明4[PF6]中存在两个等价的Cp*, 与晶体结构解析结果一致.
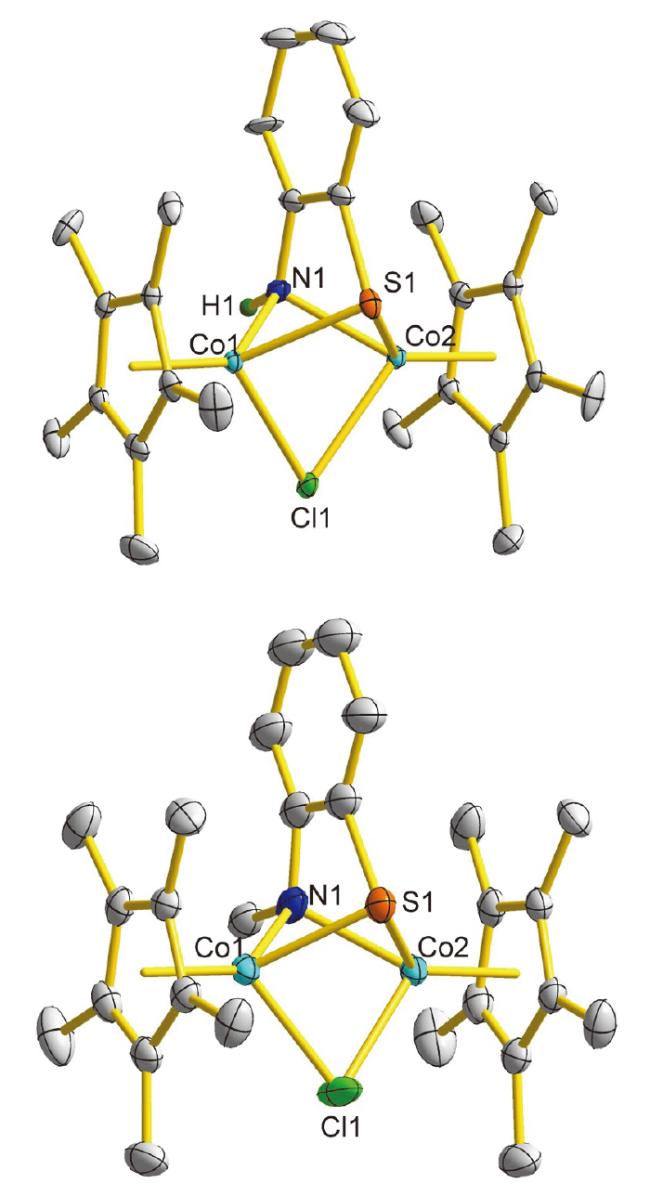
对配合物4[BPh4]的分子结构分析表明, 其Co…Co间距(0.27839(7) nm和0.2805(1) nm)均短于配合物IV (0.2969(2) nm)[15], 这归因于氮原子和氯原子较小的原子半径. 进一步对比4a[BPh4]与引入供电子基团甲基的4b[BPh4]发现: 甲基的引入导致双钴中心电子云密度升高, Lewis酸性降低, 具体表现为两个Cp*所在平面形成的二面角由6.0(3)°缩减至1.9(3)°, 同时钴中心与abtR配体的平均键长从0.2145(2) nm增至0.2156(4) nm(表2). 上述分子结构的差异表明, 供电子基团可能削弱了Co—S与Co—N的键强度, 进而影响配合物结构稳定性.
表2 配合物4a[BPh4]和4b[BPh4]的主要结构数据Table 2 Main structural data of complexes 4a[BPh4] and 4b[BPh4] |
| 4a[BPh4] | 4b[BPh4] | |
|---|---|---|
| 距离/nm | ||
| Co…Co | 0.27839(7) | 0.2805(1) |
| Co—N | 0.1992(3) | 0.204(1) |
| Co—S | 0.2298(1) | 0.2277(6) |
| Co—Cl | 0.2337(1) | 0.2276(2) |
| Co—Cp* | 0.16769(3) | 0.16875(6) |
| 二面角/(°) | ||
| Cp*1∠Cp*2 | 6.0(3) | 1.9(3) |
2.2 邻氨基苯硫酚桥联双金属配合物的反应性
钴叠氮配合物广泛应用于分子磁性[24]、药物化学[25]以及固氮等领域[26]. 本课题组在双钴叠氮化物的合成方面具有实践工作的积累. 2017年, 报道了首例硫桥联双钴叠氮桥联配合物[Cp*Co(μ-SEt)2(μ-N3)- CoCp*][BPh4] (VI)[27]. 2023年, 使用金刚烷基硫桥联双钴配合物与叠氮钠反应, 得到了双钴氮化物, 后者可逐步还原质子化产氨[28]. 然而, 上述双钴配合物与叠氮试剂反应过程中, 难捕获关键的中间体——双金属叠氮化物. 因此, 在成功制备配合物2[PF6]、3[PF6]和4[PF6]之后, 进一步考察了三种配合物与叠氮试剂的反应性, 尝试合成稳定的双金属叠氮化物.
如图5所示, 配合物4a[PF6]与1.5 equiv.的NaN3反应, 以63%产率得到硫桥联双钴叠氮桥联配合物[Cp*Co(μ-η2:η2-abt)(μ-N3)CoCp*][PF6] (5[PF6])(图6). 然而在相同条件下, 配合物4b[PF6]仅得到分解产物1b, 反应性的差异可归因于甲基的引入减弱了Co—S和 Co—N的键强度, 使双核产物不能稳定存在, 最终分解为单核配合物. 同时, 对配合物2[PF6]和3[PF6]与叠氮试剂的反应进行了研究, 然而二者均未能实现叠氮基团的配位. 值得一提的是, 2[PF6]与叠氮试剂反应发生分解, 而3[PF6]则不发生反应, 这可能由于半夹心的结构使配合物3[PF6]具有远超另外两种构型的稳定性, 难以发生转化.
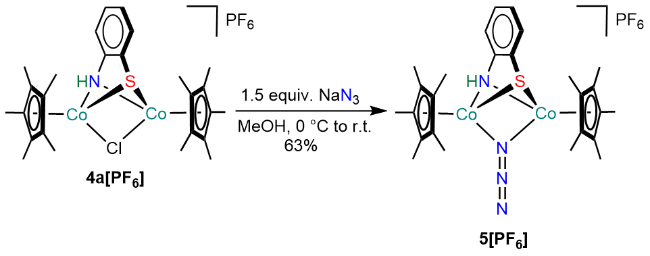
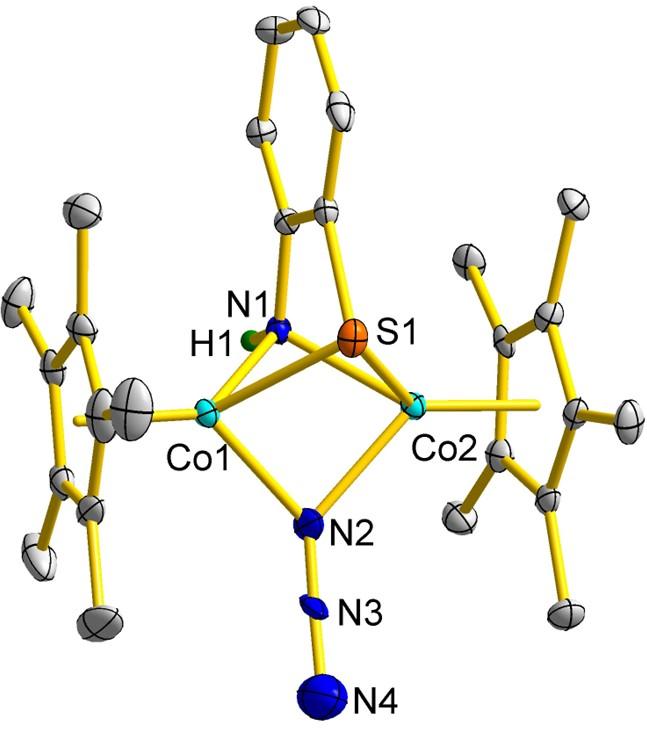
对配合物5[PF6]的分子结构研究表明, 叠氮基团以μ1,1-η1:η1的配位模式桥联于双钴中心, 构型与前体配合物4a[PF6]相同, abt仍采用μ-η2:η2配位模式. 叠氮基团的引入使两个Cp*所在平面的二面角从6.0(3)°增加到9.7(5)°. 双钴间距离从0.27839(7) nm减小到0.2751(1) nm, 为硫桥联双钴叠氮桥联配合物中最短的[29]. 此外, N2—N3—N4键角为168(1)°, 小于本课题组已报道的硫桥联双钴叠氮桥联配合物VI (176.3(8)°)[27]. 红外光谱图中2054 cm−1处的吸收峰归属为μ1,1-η1:η1型叠氮配体的伸缩振动[30].
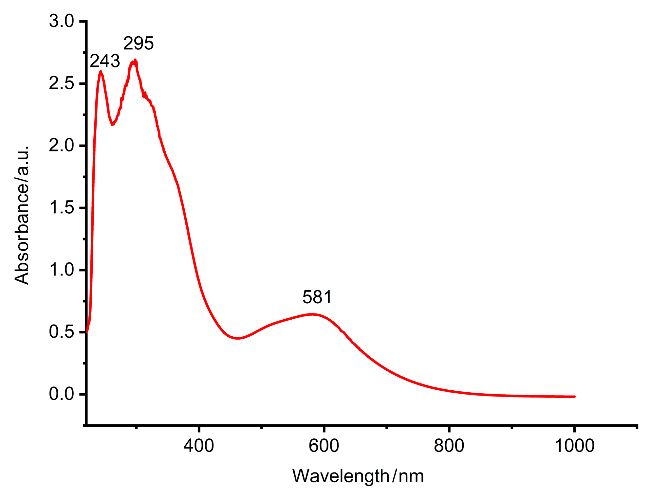
通常情况下, 叠氮配合物在光照或加热条件下不稳定, 容易脱除氮气生成对应的氮化物. 比如配合物VI在常温条件下缓慢脱除氮气, 得到氮原子插入到钴硫键之间的产物[27]. 不同的是, 配合物5[PF6]室温条件下能在溶液中稳定存在. 首先, 考察了5[PF6]在光照条件下的反应性, 为明确光解的最佳激发波长, 对5[PF6]进行紫外-可见光谱测试, 如图7所示, 紫外-可见光谱在243、295和581 nm处具有特征吸收, 将295 nm处的吸收波长归属为叠氮配体的n-π*跃迁[31]. 使用该波长的光对5[PF6]进行光照, 未发生反应. 还考察了5[PF6]在加热条件下的稳定性, 以四氢呋喃作为溶剂, 配合物5[PF6]可在60 ℃加热条件下保持稳定.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
本研究报道了一系列邻氨基苯硫酚桥联双金属配合物的合成、表征及其与叠氮钠的反应. 采用单核前体定向组装策略, 通过单核钴配合物1与单核铁、钌和钴前体反应, 合成了三种具有不同配位模式的双金属配合物2[PF6]~4[PF6]. 其中, 配合物2[PF6]和3[PF6]为罕见的邻氨基苯硫酚桥联异双核配合物. 而配合物4[PF6]中桥联的氯离子, 可以作为潜在的反应位点, 与叠氮钠反应制备首例室温下稳定的硫桥联双钴叠氮桥联配合物5[PF6]. 这一系列结构新颖的含钴的双金属配合物的成功构筑为深入开展硫桥联异/同核配合物的功能拓展提供了良好的反应平台, 同时为双金属叠氮配合物的设计合成提供了新思路.
4 实验部分
如无特别说明, 所有操作都采用标准的无水无氧Schlenk操作或在手套箱内完成, 实验所用的高纯氩气从大连化学物理研究所购买, 经脱氧柱纯化后直接使用. 实验所使用的溶剂均由PureSolv MD5溶剂处理装置纯化.
4.1 [Cp*Co(abtMe)] (1b)的制备
在室温下, 向[Cp*CoI2(CO)] (1.0 g, 2.1 mmol)的二氯甲烷(50 mL)溶液中加入abtMe (0.29 g, 2.1 mmol), 溶液颜色由紫红色变为棕红色, 随后加入三乙胺(0.59 mL, 4.2 mmol), 溶液颜色立即变为蓝色. 反应1 h后, 水洗分液, 有机相经无水硫酸镁干燥. 过滤除去干燥剂, 真空除去溶剂, 得到暗蓝色粉末[Cp*Co(abtMe)] (1b, 0.68 g, 1.99 mmol, 95%). 正己烷冷冻结晶获得适用于X-射线单晶衍射的晶体.
1H NMR (400 MHz, CDCl3, 298 K) δ: 1.97 (s, 15H, Cp*-H), 4.08 (s, 3H, NMe-H), 6.96 (t, J=6.96 Hz, 1H, abt-H), 7.11 (t, J=7.22 Hz, 1H, abt-H), 7.36 (d, J=8.52 Hz, 1H, abt-H), 7.58 (d, J=8.18 Hz, 1H, abt-H). IR (KBr) ν: 3041, 2976, 2910, 2857, 1679, 1474, 1154, 1321, 746, 541 cm−1. Anal. calcd for C17H22CoNS: C 61.62, H 6.69, N 4.23; found C 61.30, H 6.64, N 4.54.
4.2 [Cp*Co(μ-η2:η4-abtR)FeCp*][PF6] (2[PF6])的制备
在-78 ℃下, 向[Cp*Fe(MeCN)3][PF6] (50.0 mg, 1.09 mmol)的二氯甲烷(5 mL)溶液中加入1 (1a: 34.6 mg, 1.09 mmol; 1b: 37.2 mg, 1.09 mmol). 溶液逐渐由蓝色变为紫红色, 升至室温后继续反应1 h. 真空除去溶剂, 用正己烷(5 mL×3)洗涤产物, 干燥后得到黑色粉末[Cp*Co(μ-η2:η4-abtR)FeCp*][PF6] (2a[PF6], R=H, 64.7 mg, 0.99 mmol, 91%; 2b[PF6], R=Me, 66.9 mg, 1.00 mmol, 92%). 二氯甲烷/正己烷双溶剂扩散法得到适用于X-射线单晶衍射的晶体.
2a[PF6]: 1H NMR (400 MHz, CDCl3, 298 K) δ: 1.87 (s, 15H, Cp*-H), 1.42 (s, 15H, Cp*-H), 7.16 (d, J=8.69 Hz, 1H, abt-H), 7.37~7.45 (m, 2H, abt-H), 7.92 (d, J=8.62 Hz, 1H, abt-H), 8.61 (s, 1H, NH); HRMS(ESI) calcd for C26H35CoFeNS 508.1172, found 508.1168. IR (KBr) ν: 3318 (vN-H), 2936, 2922, 2854, 1651, 1471, 1381, 1261, 1023, 842, 756, 558 cm−1. Anal. calcd for C26H35CoF6FeNPS: C 47.80, H 5.40, N 2.14; found C 46.88, H 5.74, N 2.02.
2b[PF6]: 1H NMR (400 MHz, CD2Cl2, 298 K) δ: 1.78 (s, 15H, Cp*-H), 1.38 (s, 15H, Cp*-H), 4.03 (s, 3H, NMe-H), 7.35 (d, J=8.79 Hz, 1H, abt-H), 7.52 (d, J=9.08 Hz, 1H, abt-H), 7.66 (t, J=7.76 Hz, 2H, abt-H). HRMS (ESI) calcd. for C27H37CoFeNS 522.1329, found 522.1329. IR (KBr) ν: 2957, 2916, 2850, 1588, 1428, 1379, 1157, 1017, 841, 729, 556 cm−1. Anal. calcd for C27H37CoF6- FeNPS: C 48.59, H 5.59, N 2.10; found C 48.87, H 5.09, N 2.17.
4.3 [Cp*Co(μ-η2:η6-abtR)RuCp*][PF6] (3[PF6])的制备
在-78 ℃下, 向[Cp*Ru(MeCN)3][PF6] (50.4 mg, 1.0 mmol)的二氯甲烷(5 mL)溶液中加入1 (1a: 31.7 mg, 1.0 mmol; 1b: 34.1 mg, 1.0 mmol). 溶液逐渐由蓝色变为紫红色, 升至室温后继续反应1 h. 真空除去溶剂, 用正己烷(5 mL×3)洗涤产物, 干燥后得到深紫色粉末 [Cp*Co(μ-η2:η6-abtR)RuCp*][PF6] (3a[PF6], R=H, 68.5 mg, 0.98 mmol, 98%; 3b[PF6], R=Me, 67.7 mg, 0.95 mmol, 95%). 为得到适合于X-射线单晶衍射的晶体, 配合物3a[PF6]或3b[PF6]与四苯硼钠在二氯甲烷溶剂中室温反应1 h, 交换抗负离子后, 通过二氯甲烷/正己烷双溶剂扩散法得到3a[BPh4]和3b[BPh4]的单晶.
3a[PF6]: 1H NMR (400 MHz, CDCl3, 298 K) δ: 1.94 (s, 15H, Cp*-H), 1.55 (s, 15H, Cp*-H), 4.92 (t, J=5.23 Hz, 1H, abt-H), 5.01 (t, J=5.50 Hz, 1H, abt-H), 5.91 (d, J=5.47 Hz, 1H, abt-H), 5.98 (d, J=5.95 Hz, 1H, abt-H), 9.14 (s, 1H, NH). HRMS(ESI) calcd for C26H35CoRuNS 554.0872, found 554.0865. IR (KBr) ν: 3348 (vN-H), 2961, 2917, 2848, 1736, 1652, 1499, 1385, 1267, 1018, 840, 733, 558 cm−1. Anal. calcd for C26H35CoF6NPRuS: C 44.70, H 5.05, N 2.01; found C 44.77, H 5.23, N 2.15.
3b[BPh4]: 1H NMR (400 MHz, CDCl3, 298 K) δ: 1.77 (s, 15H, Cp*-H), 1.46 (s, 15H, Cp*-H), 3.77 (s, 3H, NMe-H), 4.82~4.89 (m, 2H, abt-H), 5.35 (d, J=6.06 Hz, 1H, abt-H), 5.70 (d, J=5.47 Hz, 1H, abt-H), 6.89 (t, J=6.89 Hz, 4H, Ph-H), 7.02 (t, J=7.15 Hz, 8H, Ph-H), 7.43 (s, 8H, Ph-H). HRMS (ESI) calcd for C27H37CoRuNS 568.1029, found 568.1021. IR (KBr) ν: 3361, 3054, 2918, 2854, 1881, 1579, 1478, 1428, 1293, 841, 733, 613 cm−1. Anal. calcd for C51H57BCoNRuS: C 69.07, H 6.48, N 1.58; found C 68.68, H 6.53, N 1.47.
4.4 [Cp*Co(μ-η2:η2-abtR)(μ-Cl)CoCp*][PF6] (4[PF6])的制备
在-78 ℃下, 向[Cp*Co(μ-Cl)2(t-Cl)2CoCp*] (53.0 mg, 1.0 mmol)的甲醇(5 mL)溶液中依次加入1 (1a: 31.7 mg, 1.0 mmol; 1b: 34.1 mg, 1.0 mmol)和六氟磷酸钾(18.4 mg, 1.0 mmol). 升至室温后继续反应1 h. 真空除去溶剂, 用二氯甲烷(5 mL×2)提取产物并抽干, 用正己烷 (5 mL×3)洗涤产物, 干燥后得到黑色粉末[Cp*Co(μ-η2:η2-abtR)(μ-Cl)CoCp*][PF6] (4a[PF6], R=H, 56.7 mg, 0.82 mmol, 82%; 4b[PF6], R=Me, 66.3 mg, 0.94 mmol, 94%). 为得到适合于X-射线单晶衍射的晶体, 配合物4b[PF6]与四苯硼钠在二氯甲烷溶剂中室温反应1 h, 交换抗负离子后, 通过二氯甲烷/正己烷双溶剂扩散法得到4b[BPh4]的单晶.
4a[PF6]: 1H NMR (400 MHz, CDCl3, 298 K) δ: 1.15 (s, 30H, Cp*-H), 5.65 (br, 2H, abt-H), 6.86 (br, 2H, abt-H). HRMS(ESI) calcd for C26H35Co2NS 546.0843, found 546.0838. IR (KBr) ν: 3312 (vN-H), 3064, 2987, 2914, 1564, 1472, 1377, 1020, 841, 730, 557 cm−1. Anal. calcd for C26H35Co2F6NPS: C 45.13, H 5.10, N 2.02; found C 45.28, H 5.42, N 1.95.
4b[BPh4]: 1H NMR (400 MHz, CDCl3, 298 K) δ: 0.94 (s, 30H, Cp*-H), 2.64 (s, 3H, NMe-H), 6.35 (br, 1H, abt-H), 6.52 (br, 1H, abt-H), 6.67 (br, 2H, abt-H) 6.88 (br, 4H, Ph-H), 7.03 (br, 8H, Ph-H), 7.41 (br, 8H, Ph-H). HRMS(ESI) calcd. for C27H37ClCo2NS 560.0999, found 560.0995. IR (KBr) ν: 3054, 2982, 2913, 1941, 1580, 1375, 1267, 1016, 630, 611 cm−1. Anal. calcd for C51H57BCoNRuS: C 69.59, H 6.53, N 1.59; found C 69.84, H 6.67, N 1.15.
4.5 [Cp*Co(μ-η2:η2-abt)(μ-N3)CoCp*][PF6] (5[PF6])的制备
在0 ℃下, 向4a[PF6] (50.0 mg, 0.72 mmol)的甲醇(5 mL)溶液中加入叠氮钠(7.1 mg, 1.08 mmol). 升至室温后继续反应1 h. 真空除去溶剂, 用二氯甲烷(5 mL×2)提取产物并抽干, 用正己烷(5 mL×3)洗涤产物, 干燥后得到黑色粉末[Cp*Co(μ-η2:η2-abt)(μ-N3)CoCp*][PF6] (5[PF6], 20.0 mg, 0.45 mmol, 63%). 通过二氯甲烷/正己烷双溶剂扩散法得到5[PF6]的单晶.
5[PF6]: 1H NMR (400 MHz, CDCl3, 298 K) δ: 1.23 (s, 30H, Cp*-H), 7.00 (d, J=7.6 Hz, 1H, abt-H), 7.08 (d, J=8.4 Hz, 1H, abt-H), 7.16 (d, J=7.46 Hz, 1H, abt-H), 7.77 (d, J=6.00 Hz, 1H, abt-H). HRMS(ESI) calcd for C26H35Co2N4S 553.1246, found 553.1243. IR (KBr) ν: 3317 (vN-H), 2921, 2054 (vN꞊N), 1466, 1385, 1261, 1021, 840, 736, 558 cm−1. Anal. calcd for C26H35Co2F6N4PS: C 44.71, H 5.05, N 8.02; found C 45.33, H 4.95, N 7.41.
支持信息(Supporting Information) 所合成的配合物的氢核磁共振谱图、红外谱图及晶体数据等材料可以免费从本刊网站上下载. 配合物的晶体结构数据已上传至英国剑桥晶体数据库(Cambridge Crystallographic Data Centre), CCDC号码为2402081、2402082、2402083、2402084、2417208、2417209、2417210和2417465.
(Cheng, B.)