


1 引言
作为分子结构修饰的重要策略, 向有机分子中引入含氟官能团能够显著优化母体分子的理化性质与生物活性[1]. 在众多含氟官能团中, 氟烷基凭借其独特的强电负性与优异亲脂性, 在改善药物分子的脂溶性、化学稳定性及代谢稳定性方面呈现出独特的优势, 因此氟烷基化技术在药物研发和生命科学领域发挥着重要的作用[2]. 光催化自由基氟烷基化反应具有官能团耐受性高和反应条件温和等优点, 受到了科研工作者的广泛关注[3]. 目前, 研究者已开发出多种方法[4], 利用不同试剂, 如氟烷基卤化物[5]、氟烷基羧酸[6]、氟烷基亚磺酸钠[7]等, 向各类分子骨架中引入氟烷基. 这些试剂通过提供温和高效的C—CF3键构建途径, 展现出独特的优势. 其中, 氟烷基亚磺酸钠作为一种廉价易得、性质稳定的氟烷基化试剂, 被用于多种氟烷基化反应[8].
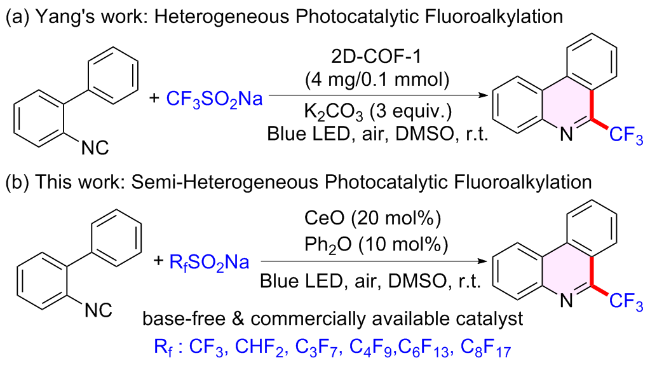
可见光催化通过可见光照激发电子来引发化学反应, 能在温和条件下实现化学键的断裂和重组, 具有绿色环保、条件温和以及官能团耐受性好等优点[13]. 经过科研工作者不懈的努力, 一系列可见光催化反应体系被发现, 并成功应用于复杂分子的合成中[14]. 可见光催化2-异腈基联苯与氟烷基试剂的自由基氟烷基化反应得到了一定的发展[15]. 其中, 以廉价易得的氟烷基亚磺酸钠为氟烷基源的可见光催化2-异腈基联苯自由基氟烷基化反应备受关注[16]. 均相可见光催化2-异腈基联苯氟烷基化反应需要使用不易回收的有机小分子光催化剂/氧化剂组合[17]或一定量的光促进剂[18], 导致反应成本增加和环境负担加重. 2019年, 杨晓博等[19]利用新型二维共价有机骨架材料作为多相光催化剂, 首次实现6-氟烷基菲啶化合物的多相光催化合成. 然而, 该反应使用的共价有机骨架材料光催化剂的制备过程繁琐, 还需使用大量的无机碱为添加剂, 影响了反应的实用性(Scheme 1a). 因此, 开发绿色实用的6-氟烷基菲啶的可见光催化合成方法仍具有重要的研究价值.
2 结果与讨论
2.1 二苯醚介导氧化铈半多相光催化2-异腈基联苯氟烷基化反应
2.1.1 条件优化
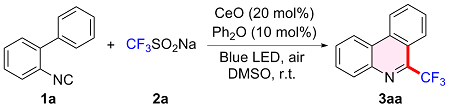
最初, 我们选择2-异腈基联苯(1a)和三氟甲基亚磺酸钠(CF3SO2Na, 2a)作为模型底物来优化反应条件(表1). 经过一系列条件筛选后, 发现以二甲基亚砜(DMSO)作为溶剂, 氧化铈(CeO2)作为半导体光催化剂、二苯醚(Ph2O)作为氧化还原介体, 在常温空气氛围下经10 W蓝光LED照射进行氟烷基化反应24 h, 目标产物3aa的气相色谱(GC)产率可达73% (Entry 1). 对比实验表明: 更换其它半导体光催化剂未提升产率(Entries 2~3). 在没有光催化剂的情况下, 反应不能发生(Entry 4). 氧化还原催化剂的筛选显示, 使用其他氧化还原催化剂替代二苯醚会导致催化效率显著下降(Entry 5). 不使用任何氧化还原介体时, 光催化反应效率大幅降低, 表明氧化还原介体在反应中发挥着关键性作用(Entry 6). 光源优化实验显示蓝色LED是本反应的最优光源(Entries 7~9). 使用12 W的蓝色LED作为光源, 目标产物3aa产率没有提高; 而使用8 W的蓝色LED作为光源会导致3aa的产率降低(Entry 10). 溶剂体系考察发现, 使用丙酮(Acetone)、乙酸乙酯(EtOAc)和N,N-二甲基甲酰胺(DMF)作为溶剂, 3aa产率明显降低(Entry 11). 氮气氛围或避光条件下反应完全被抑制(Entry 12), 凸显该光催化体系对氧气及特定光源的依赖性.
表1 反应条件优化aTable 1 Optimization of reaction conditionsa |
| Entry | Varying from the standard conditions | Yieldb/% |
|---|---|---|
| 1 | None | 73 |
| 2 | g-C3N4, Fe2O3, BiVO4 instead of CeO2 | 36, 45, 32 |
| 3 | CdO, MoO2, ZnO instead of CeO2 | 41, 34, 38 |
| 4 | Without semiconductor photocatalyst | N.R. |
| 5 | NHPI, Cp2Fe, Ph3N instead of Ph2O | 41, 28, 31 |
| 6 | Without redox catalyst | 33 |
| 7 | Purple LED instead of blue LED | 39 |
| 8 | Green LED instead of blue LED | N.R. |
| 9 | White LED instead of blue LED | N.R. |
| 10 | LED (12 W), LED (8 W) instead of LED (10 W) | 72, 63 |
| 11 | Acetone, EtOAc, DMF instead of DMSO | 21, trace, 50 |
| 12 | Without air or light | N.R. |
a Condition: 1a (0.1 mmol), 2a (0.2 mmol), CeO2 (20 mol%), Ph2O (10 mol%), DMSO (2 mL), air, blue LED (10 W), 24 h under room temperature. b Estimated by GC using dodecane as an internal reference. |
2.1.2 底物扩展
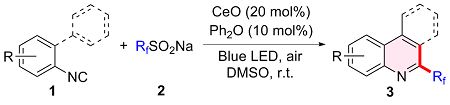
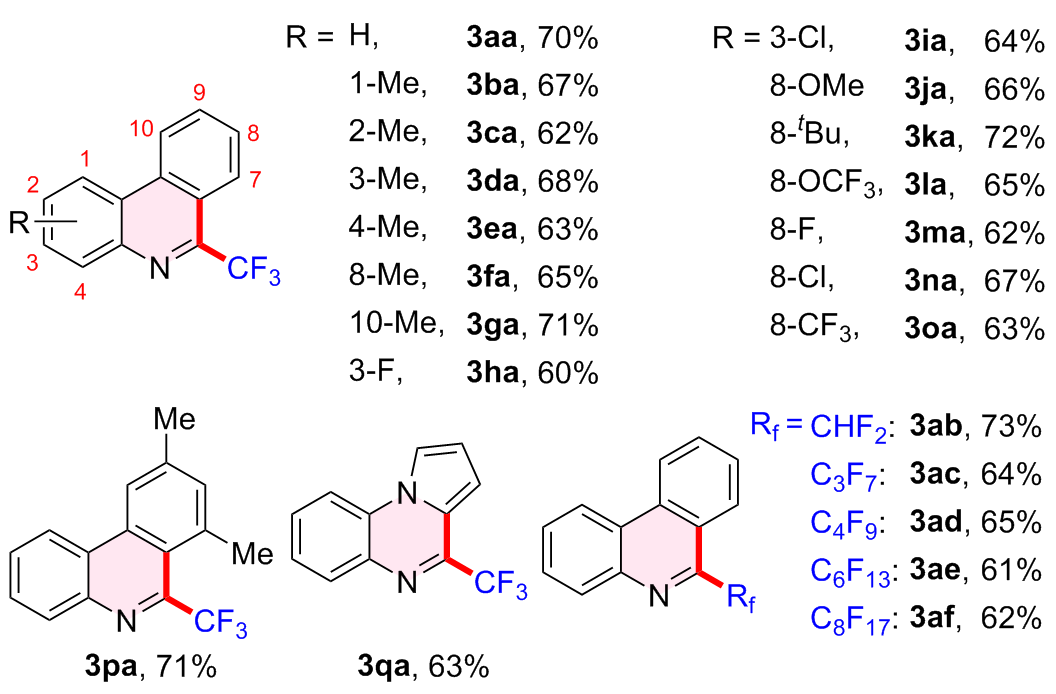
在获得了最优反应条件(表1, Entry 1)之后, 我们对可见光诱导2-异腈基联苯氟化反应的底物普适性进行了探究(表2). 2-异腈基联苯的苯环上取代基的位置对于烷基化反应产率的影响不大, 能以60%~72%的分离产率得到目标产物3aa~3oa. 苯环上无论是取代有供电子基团或吸电子基团的2-异腈基联苯衍生物都可以很好地兼容该反应体系, 以良好到优秀的分离收率得到目标产物. 其中, 两种二取代异腈化物均成功参与反应, 分别以71%和63%的产率获得对应产物3pa和3qa. 紧接着对氟代亚磺酸钠底物进行了普适性考察. CHF2- SO2Na在标准条件下与1a反应顺利, 得到产物3ab, 产率为73%. 全氟烷基亚磺酰钠, 包括C3F7SO2Na、C4F9SO2Na、C6F13SO2Na和C8F17SO2Na有效地进行了氟烷基化反应, 并以良好的收率获得3ac~3af的目标产物. 使用CF3SO2Cl替代CF3SO2Na时, 反应不能发生.
表2 2-异腈基联苯范围aTable 2 Scope of 2-isocyanobiaryla |
 |
a Condition: 1 (0.2 mmol), 2 (0.4 mmol), Ce2O (20 mol%), Ph2O (10 mol%), DMSO (2 mL), air, blue LED (10 W), 24 h under room temperature. |
2.2 放大实验
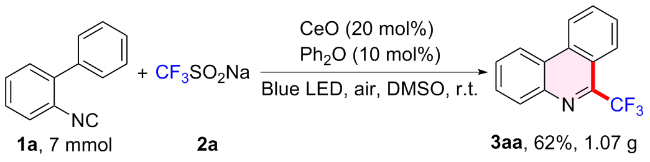
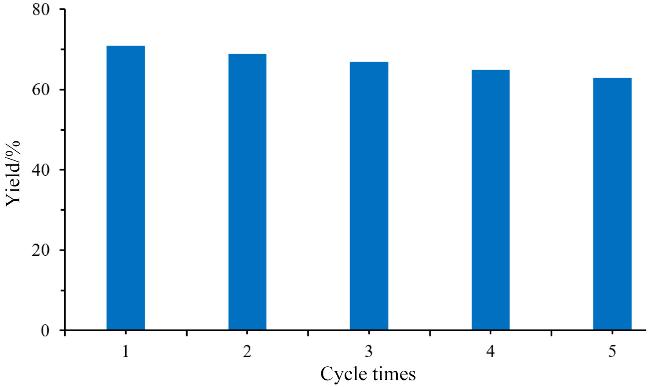
2.3 控制反应
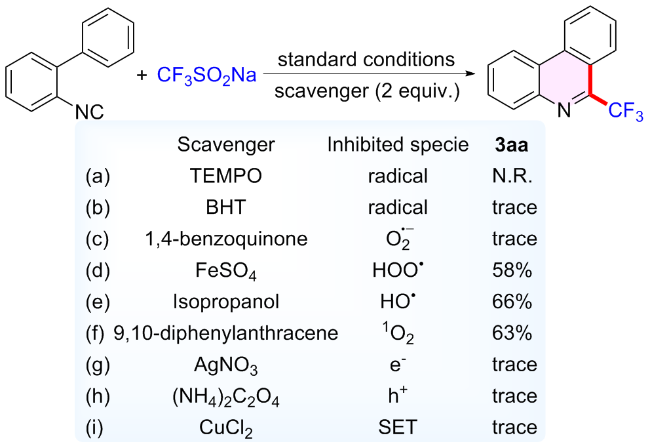
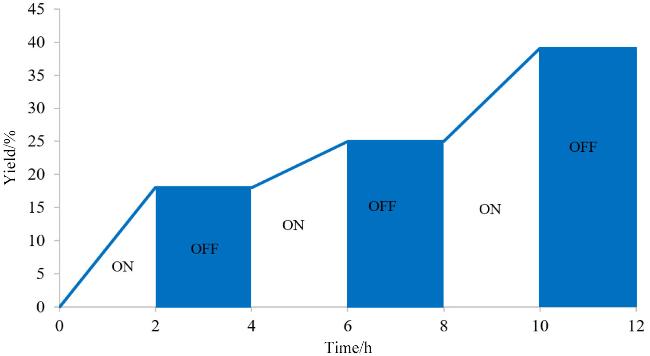
随后对反应的机理进行了探究(Scheme 3), 在标准条件下向反应中加入2 equiv.的自由基抑制剂四甲基哌啶氧化物(TEMPO)或2,6-二叔丁基对甲酚(BHT), 反应被完全抑制(Scheme 3a和3b). 在反应中, 加入超氧自由基阴离子($\text{O}_{2}^{\bullet }$)抑制剂1,4-苯醌, 只检测到了痕量的产物3aa (Scheme 3c), 表明$\text{O}_{2}^{\bullet }$在该反应历程中发挥着关键性作用; 加入过氧化自由基(HOO•)抑制剂FeSO4, 单线态氧(1O2)抑制剂, 9,10-二苯基蒽(9,10-diphenyl-anthracene)、羟基自由基(HO•)抑制剂异丙醇则对该反应没有显著的影响(Scheme 3d~3f). 当添加AgNO3或(NH4)2C2O4到该反应中时, 仅产生微量的3aa, 这表明光生电子(e-)和光生空穴(h+)对于非均相光催化过程都是必需的(Scheme 3g~3h). 此外, 向模型反应中加入单电子转移抑制剂CuCl2完全抑制了3aa的形成, 证明反应可能包括单电子转移过程(Scheme 3i). 光照开/关实验结果排除了该反应是自由基链式反应的可能性(图2).
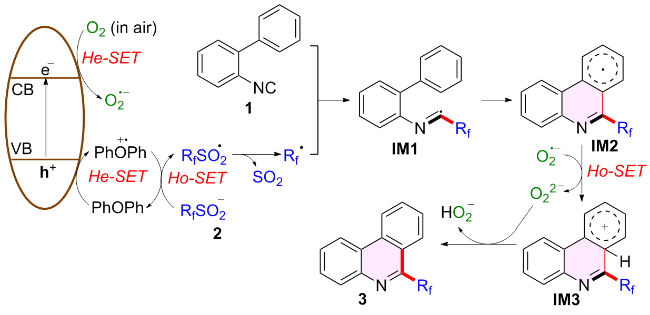
基于上述机理实验结果和相关文献报道[17,19], 我们提出了该半多相光催化反应可能的反应机理(Scheme 4). 在蓝色LED灯照射下, Ce2O半导体吸收光子被激发产光生电子-空穴对. 光生电子通过多相单电子转移过程(He-SET)将空气中的分子氧还原为活性更高的超氧自由基阴离子($\text{O}_{2}^{\bullet }$). 同时, 光生空穴通过多相单电子转移氧化二苯醚生成氧化态的二苯醚自由基阳离子, 其通过均相单电子氧化(Ho-SET)氟烷基亚磺酸钠2生成氟烷基砜基自由基${{R}_{f}}S\text{O}_{2}^{\bullet }$, 随后迅速脱SO2生成氟烷基自由基$R_{f}^{\bullet }$. $R_{f}^{\bullet }$进攻2-异腈基联苯1的异腈基生成亚胺自由基中间体IM1, 其发生分子内环化得到环闭合自由基中间体IM2. $\text{O}_{2}^{\bullet }$单电子氧化中间体IM2生成超氧阴离子($\text{O}_{2}^{2}$)[22] 和菲啶阳离子中间体IM3, 其发生脱质子芳构化过程生成目标产物3.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
本文报道了一种以廉价易得的本征氧化铈半导体作为多相光催化剂, 二苯醚作为均相氧化还原助催化剂, 氟烷基亚磺酸钠作为自由基氟烷基化试剂, 无需强氧化剂和碱添加剂的二苯醚介导氧化铈半多相光催化2-异腈基联苯氟烷基化反应, 以60%~73%的收率合成了22种6-氟烷基菲啶化合物. 该半多相光催化合成方法具有反应条件温和、官能团耐受性好、实验操作简单等优点, 为6-氟烷基菲啶化合物的制备提供了一种绿色可持续的新方法.
4 实验部分
向装有磁力搅拌子的10 mL Schlenk烧瓶中加入1 (0.2 mmol)、2 (0.4 mmol)、氧化铈(0.04 mmol)、二苯醚(0.02 mmol)和二甲基亚砜(2 mL). 然后搅拌反应混合物, 在环境温度和空气气氛下用10 W蓝色LED照射, 通过薄层色谱分析监测反应过程. 反应完成后, 通过过滤去除固体. 减压蒸发滤液, 通过硅胶色谱柱获得纯产物3.
(Cheng, F.)