


1 引言
含氮杂环化合物作为众多天然产物、药物分子和功能化合物中普遍存在的结构单元, 因其显著的生物学特性与药理学价值而备受关注(图1)[1]. 这类化合物在有机合成化学、农药开发及药物研发领域具有广泛应用[2]. 其中, 吲哚酮衍生物因其多样化的生物活性成为研究热点[3], 包括抗菌[1]、抗病毒[4]、抗白血病[4]、镇痛[5]、抗心力衰竭[5]、抗人类免疫缺陷病毒(HIV)[1]、抗癌[6]及抗类风湿性关节炎[7]等特性. 鉴于吲哚酮的广泛生物活性和丰富的合成效用, 该骨架的构建仍然是合成有机化学的一个重要目标. 近年来, 合成吲哚酮的一般方法包括分子内成环反应[8], 以及在吲哚酮骨架上对C-2位、C-3位或N位进行官能团修饰, 以引入特定官能团[9].
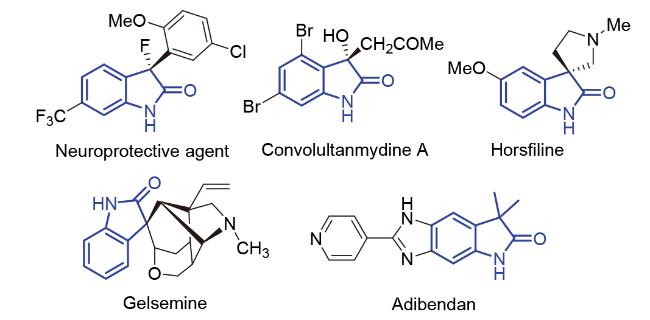
N-芳基丙烯酰胺因其易合成、产率高等特性, 成为构建吲哚酮骨架的优质底物之一. 合成吲哚酮的传统方法主要包括以下两类: 第一类为脱卤环化反应(Scheme 1~a)[10], 该策略通过邻位卤代的N-芳基丙烯酰胺(X=Cl、Br、I)在Pd、Ni、Rh、Ir、Fe等过渡金属催化下实现 C—X键的活化及分子内环化; 另一类为热驱动过渡金属催化以及近年来兴起的过渡金属/可见光双催化体系, 通常采用Pd、Ag或Cu等金属活化C(sp2)—H键(Scheme 1~b)[11], 实现N-芳基丙烯酰胺的直接分子内偶联. 虽然这类方法避免了预先官能化步骤, 但仍存在显著局限性: 底物需预先进行邻位官能团修饰; 高负载量的贵金属催化剂可能导致药物残留污染; 反应后处理操作复杂; 通常需要苛刻的热力学条件(高温)或特殊配体.
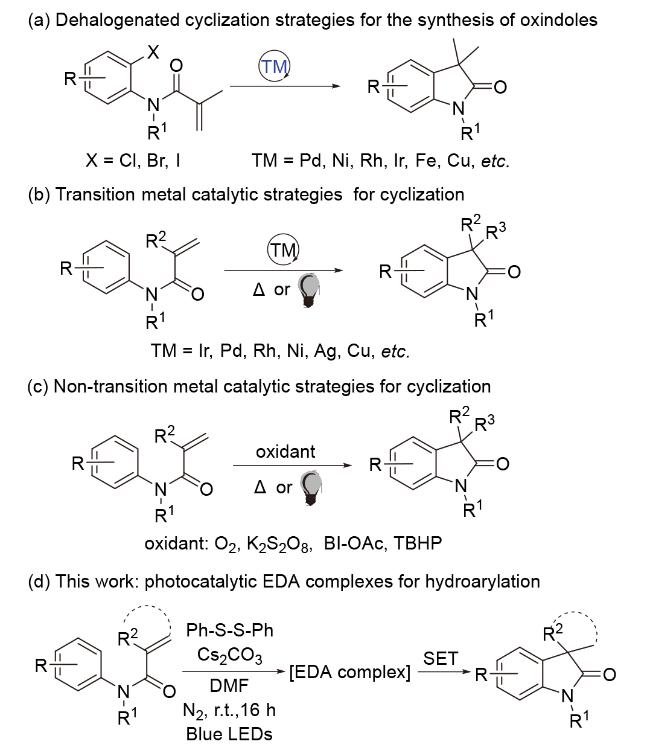
为突破上述限制, 开发绿色、无过渡金属参与的催化体系成为当前研究的重要方向. 氧化环化策略通过氧化剂[如O2、K2S2O8或叔丁基过氧化氢(TBHP)]介导引发电子转移的环化反应, 完全避免了过渡金属的参与(Scheme 1~c)[12]. 这一特点虽然规避了过渡金属催化体系存在的残留污染问题, 但是其反应产率与选择性仍受限于氧化剂的性质: 化学计量需求高, 导致成本增加及后处理负担加重; 强氧化性易引发底物过度官能化或分解, 导致底物普适性较差等.
为突破上述限制, 开发绿色、无过渡金属参与的催化体系成为当前研究的重要方向. 电子供体-受体(EDA)复合物策略进行的化学反应, 除了光作为外部能量来源外, 其反应条件温和, 无需外加强氧化剂或过渡金属催化剂, 仅依赖光能与分子间弱相互作用即可实现转化[13-17]. 这一特性不仅降低了反应能耗与污染风险, 还拓宽了底物的适用范围. 尽管吲哚酮骨架的合成方法已较为丰富, 但基于EDA复合物的光催化策略在N-芳基丙烯酰胺环化这一领域的应用研究尚不多见. 在这种情况下, 我们设计了一种可见光诱导的硫负离子形成EDA复合物的新策略(Scheme 1~d). 在Cs2CO3提供的弱碱性条件下, PhSSPh发生微量水解生成的PhS⁻作为强电子供体, 与N-芳基丙烯酰胺底物(电子受体)形成EDA复合物. 在可见光照射下, 该EDA复合物被活化, 驱动分子内环化高效构建多种吲哚酮骨架. 该策略在室温条件下进行, 无需额外的光催化剂、强酸或氧化剂, 操作简便, 为吲哚酮衍生物的绿色合成提供了一条新途径.
2 结果与讨论
2.1 反应条件优化
表1 反应条件优化Table 1 Conditions optimization |
| Entry | Variations from standard conditions | Yieldb/% |
|---|---|---|
| 1 | none | 56 |
| 2 | A2 instead of A1 | trace |
| 3 | A3 instead of A1 | 35 |
| 4 | A4 instead of A1 | n.r. |
| 5 | A5 instead of A1 | 25 |
| 6 | Cs2CO3 instead of K2CO3 | 70 |
| 7 | DABCO instead of K2CO3 | trace |
| 8 | DBU instead of K2CO3 | 45 |
| 9 | DIPEA instead of K2CO3 | 35 |
| 10 | H2O instead of MeCN | n.r. |
| 11 | DMF instead of MeCN | 90 |
| 12 | DMSO instead of MeCN | trace |
| 13 | 390~395 nm or white light | n.r. |
| 14 | air instead of N2 | 55 |
| 15 | O2 instead of N2 | n.d |
| 16 | without A1 or Cs2CO3 or light irradiation | n.r. |
a Reaction conditions: 1a (0.2 mmol), sulfur anion precursors (0.3 mmol) and base (0.2 mmol) in solvent (2 mL) at room temperature for 16 h under N2, 18 W blue LED (λ=455~460 nm). b Isolated yields. |
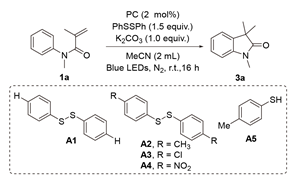
硫负离子前体结构研究表明: 给电子基取代物A2仅得痕量产物(表1, Entry 2). 吸电子基取代物A3/A4使产率分别降至35%及完全抑制(表1, Entries 3~4). 苯硫酚A5因缺乏二硫键致产率锐减(表1, Entry 5). 该现象表明给电子基阻碍S—S键均裂, 吸电子基削弱硫原子的供电子能力, 二硫键缺失则破坏自由基链循环. 碱源筛选发现: Cs2CO3提升产率至70%(表1, Entry 6), 其优势源于(1) Cs+大离子半径增强碳酸盐溶解度, 提高OH⁻浓度促进PhSSPh水解. (2)弱碱性环境抑制PhS⁻质子化, 维持活性中间体浓度. 有机碱体系[1,4-二氮杂双环[2.2.2]辛烷(DABCO)/1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)/N,N-二异丙基乙胺(DIPEA)]因碱性失配导致效率低下(表1, Entries 7~9). 溶剂效应显示: N,N-二甲基甲酰胺(DMF)以90%产率最优(表1, Entry 11), 其中等极性与非质子特性协同促进PhS-解离与EDA复合物稳定. 水体系完全抑制反应(表1, Entry 10), 质子环境破坏复合物形成. 二甲基亚砜(DMSO)因强溶剂化效应仅得痕量产物(表1, Entry 12). 相同功率下紫光(390~395 nm)与白光均不能驱动反应进行(表1, Entry 13).
综上所述, 该反应最佳条件为: 在氮气氛围下, 以PhSSPh (0.3 mmol)为硫负离子前体试剂、Cs2CO3 (0.2 mmol)作为碱, DMF (2 mL)为溶剂, 在18 W蓝光LED (λ=455~460 nm)下反应16 h, 产物3a反应产率达90%.
2.2 底物拓展
在确定了最优反应条件后, 首先系统考察了N-芳基丙烯酰胺的底物范围(表2). 该转化过程可与多种官能团兼容. 具体来说, 在苯环对位引入甲基(Me)、甲氧基(OMe)等供电子基团的底物均高效转化为吲哚酮衍生物3b~3c. 当引入卤素取代基(氟、氯、溴)时, 以75%~84%收率获得产物3d~3f. 含强吸电子基团(CF3、PhO、OCF3、CN、CO2Me)的N-芳基丙烯酰胺在该体系下同样兼容, 得到环化产物3g~3k. 这些基团广泛存在于众多药物分子或生物活性化合物中. 值得注意的是, 当苯环邻位引入Me或Cl时, 反应活性显著降低, 仅以50%和53%的中等产率获得产物3l和3m, 这可能是由于邻位取代的空间位阻所致的. 有趣的是, 当苯环的间位Me取代时, 表现出独特的区域选择性, 生成α/β区域异构体3n与3n' (比例2.3∶1), 总收率88%. 带有双取代基的苯胺和空间拥挤的萘环仍表现出良好的反应活性, 分别生成的主要产物为3o和3p. 值得说明地是, 电子中性的吡啶基丙烯酰胺底物同样实现了环化转化, 以54%的收率得到产物3q, 这一现象表明该策略对杂环体系具有潜在应用价值.
表2 光催化N-芳基酰胺的底物范围Table 2 Substrate scope of photocyclization of N-arylacrylamides |
| |

a Reaction conditions: N-arylacrylamides (0.2 mmol), PhSSPh (0.3 mmol), and Cs2CO3 (0.2 mmol) in DMF (2 mL) at room temperature for 16 h under N2, 18 W blue LED (λ=455~460 nm). b Isolated yields. |
系统考察了苯胺N-取代基的兼容性, 研究表明, 不管是大位阻异丙基和环己基还是具有电子效应的苄基和酯基均能高效转化为相应的吲哚酮4a~4d, 产率达82%~88%, 这表明N-取代基的空间和电子效应对反应结果影响较小. 此外, 四氢喹啉以及其他环状二芳基N-酰胺展现突出优势, 以64%~70%的产率顺利获得目标产物4e~4g, 证实了该反应对刚性结构底物的良好适配性. 区域选择性研究发现: 4-溴苯基、4-甲苯基和萘基底物同样产生α/β混合物(4i~4k, 结构经NMR以及前人的报道确认)[17]. 最后, 在N-芳基丙烯酰胺的α位引入烯丙基取代基以及环状烯烃, 这些底物同样能够顺利实现环化反应, 以67%~88%的产率实现环化得到产物4l~4n. 值得注意的是, 该方法在游离N-H丙烯酰胺底物的环化反应中表现出明显的局限性, 不能得到目标产物.
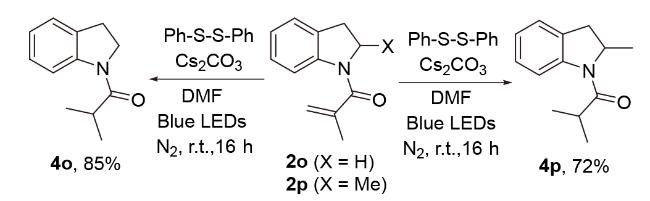
让人意外的是, 当使用吲哚啉衍生物作为底物时(Scheme 2), 可能由于环张力较大以及五元螺环稳定性差, 以中等至优异的产率得到相应的氢化产物4o和4p.
为明确反应引发机制中EDA复合物的存在, 我们进行了UV-Vis光谱实验系统分析了在0.1 mol/L下DMF体系中各组分的吸收特性(图2~a). 结果显示, 单体1a和A1在可见光区均无吸收, 1a与Cs2CO3的二元体系仅在370 nm处出现微弱吸收, 而三元体系(1a+PhSSPh+Cs2CO3)则在450~500 nm处呈现出显著的宽吸收带. 此现象初步表明在弱碱性条件下, PhSSPh水解产生的PhS⁻可能与受体1a发生相互作用, 形成具有光活性的新物种, 该物种在可见光区展现出区别于单组分及二元体系的特征吸收.
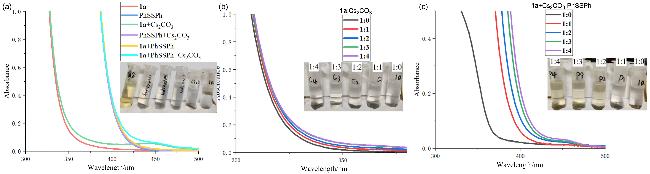
为验证此推测并探究吸收带来源, 进一步设计了对照实验: 固定1a浓度(0.01 mol/L)并递增Cs2CO3比例[n(1a)∶n(Cs2CO3)=1∶0~1∶4)时(图2~b), 仅观察到吸收带微弱红移且体系颜色无明显变化, 此现象可能来源于碱对底物的微扰; 反之, 固定1a (0.01 mol/L)和Cs2CO3 (0.01 mol/L)浓度, 梯度增加PhSSPh浓度[n(1a)∶n(PhSSPh)=1∶0~1∶4]时(图2~c), 吸收带边发生显著红移, 尾部延伸到可见光区域, 且反应体系颜色随PhSSPh浓度增加而逐渐加深. 该浓度梯度依赖性实验直接证实: (1) PhSSPh在弱碱作用下生成的PhS⁻与1a确实形成了EDA复合物; (2)该复合物在可见光区产生的新吸收带是其发生光驱动SET过程的必要条件; (3)吸收带红移程度与PhSSPh浓度呈正比.
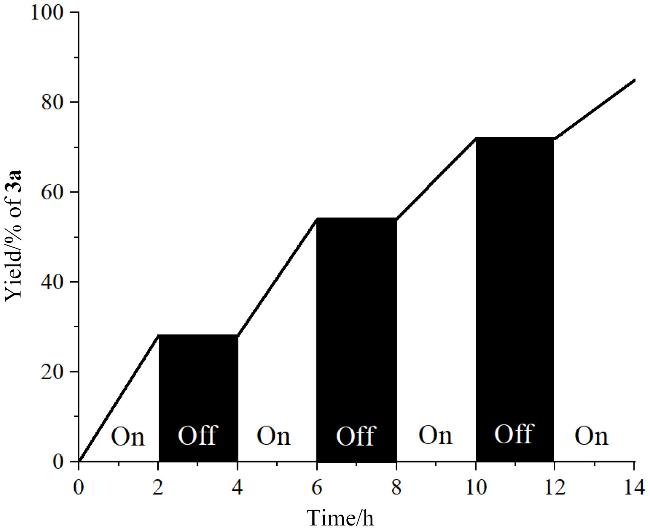
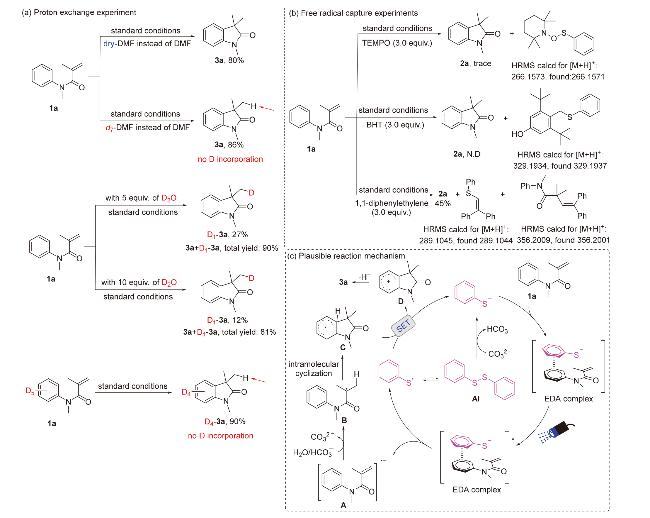
为探究光在反应中的作用, 我们进一步开展了开/关灯循环实验(图3), 结果表明: 当开灯时反应高效进行, 而中途关闭光源, 产物则不再生成, 反应停止; 重新打开灯, 反应又可继续进行. 该现象证明了反应依赖持续的可见光照射, 并非光引发的自由基链式反应, 这也与前人报道的EDA复合物光活化机制一致. 此外, 在持续蓝光照射下, 0~4 h内产率从0%急速上升至54%, 表明光活化EDA复合物可快速引发自由基环化. 为阐明质子转移机制中的质子来源, 我们设计了一系列控制实验(Scheme 3~a): 首先使用超干的DMF替代普通的DMF进行反应, 观察到产率由90%显著降低至80%, 该现象表明微量水分子参与质子转移; 进一步采用全氘代DMF (d7-DMF)时, 产物3a的1H NMR未检测到氘代信号, 排除溶剂供质子可能. 随后进行的重水(D₂O)标记实验来进一步证实水的直接参与. 当在标准反应条件下加入5 equiv. D2O时, 结果表明, 该条件下反应总产率维持不变, 但通过1H NMR检测到氘代产物D1-3a的产率为27%, 而未氘代的产物3a产率为66%, 直接证明D2O作为氘源参与质子转移; 当D2O增至10 equiv., 总产率降至81%且氘代比例仅为12%, 结合水溶剂体系反应完全抑制现象, 表明过量水会破坏EDA复合物之间的相互作用, 削弱电子转移效率. 关键性证据来自全氘代底物D5-1a实验: 反应生成90%四氘代产物D4-3a, 未检测到氘代转移产物, 明确排除芳环C(sp2)—H键贡献质子的可能性. 上述数据共同证实: 体系微量水是质子转移的唯一来源, 且适量水分子促进质子转移, 过量水则因破坏电子转移路径导致反应抑制.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
为验证反应是否经历自由基过程以及捕获关键自由基中间体, 我们在标准反应条件下向反应体系中分别加入3.0 equiv.的2,2,6,6-四甲基哌啶氧化物(TEMPO)、2,6-二叔丁基-4-甲基苯酚(BHT)及1,1-二苯乙烯作为自由基捕获剂, 实验结果表明, 反应均被显著抑制. 通过HRMS均检测到了相应的自由基捕获物的特征信号, 进一步证实该反应遵循自由基反应路径(Scheme 3~b).
根据实验数据与以往报道的文献研究[18-20], 我们提出了合理反应机理(Scheme 3~c). 在Cs2CO3提供的弱碱性条件下, A1发生微量水解生成PhS-. 该PhS-物种与N-芳基丙烯酰胺底物1a形成EDA复合物. 紫外光谱研究显示, 随着A1浓度的增加, 观测到的红移现象显著增强, 此步骤为光反应引发的关键步骤. 在可见光照射下, 该EDA复合物受激发发生SET过程, 解离生成PhS•和自由基阴离子中间体A. 同时, 体系中游离的PhSSPh在光照下发生S—S键均裂, 持续补充PhS•自由基. A则与体系中的微量水或碳酸氢根离子($\mathrm{HCO}_{3}^{-}$)发生质子化, 形成α-酰胺烷基自由基B, 随后B经历5-exo-trig分子内环化生成自由基中间体C. 中间体C在碱性环境下经单电子转移形成中间体D, D经过去质子化作用, 生成最终产物3a, 完成催化循环. 整个过程通过“微量PhS⁻引发-EDA光活化-自由基链式增长”的协同机制, 有效克服电子转移能垒, 驱动反应高效进行.
3 结论
本文开发了一种无催化剂、可见光驱动的策略, 利用EDA复合物实现N-芳基丙烯酰胺的分子内氢芳基化反应, 高效构建吲哚酮骨架. 该策略无需外加光催化剂、酸或氧化剂. 机理研究表明, 在Cs2CO3作用下, hSSPh水解产生的PhS-作为电子供体, 与底物形成光活性EDA复合物, 吸收可见光驱动单电子转移完成环化. 反应条件温和, 底物普适性良好, 兼容多种电性取代基, 操作简单, 为吲哚酮合成提供了一条绿色高效途径.
4 实验部分
4.1 N-芳基丙烯酰胺的制备
使用苯胺或甲基丙烯酰氯化合物来合成N-芳基丙烯酰胺. 首先, 向一个装有磁力搅拌棒的250 mL圆底烧瓶中加入苯胺S1 (1.0 g, 1.0 equiv.)的CH2Cl2溶液(60 mL)和三乙胺(2.0 equiv.). 将混合物在0 ℃下搅拌, 并在氮气保护下逐滴加入甲基丙烯酰氯S2a (1.5 equiv.). 待反应混合物升温至室温后, 继续搅拌12 h. 随后, 加入水(150 mL)以淬灭过量的酰氯. 将混合物转移至分液漏斗中, 分离有机层, 用饱和食盐水洗涤(100 mL×3), 并用MgSO4干燥. 减压蒸馏除去溶剂后, 最后经硅胶柱层析[200~300目, 洗脱剂V(石油醚)∶V(乙酸乙酯)=10∶1~8∶1梯度洗脱]纯化粗产物.
4.2 氢芳基化的一般程序
以底物1a的环化为例, 具体操作如下: 在干燥的反应瓶中, 加入N-芳基丙烯酰胺1a (35.0 mg, 0.2 mmol, 1.0 equiv.)、PhSSPh (65.5 mg, 0.3 mmol, 1.5 equiv.)、Cs2CO3 (71.2 mg, 0.2 mmol, 1.0 equiv.)溶于DMF (2.0 mL)中. 反应体系持续通N2鼓泡20 min, 在18 W蓝光LED (λ=455~460 nm)照射下搅拌反应16 h. 反应完成后, 用饱和氯化钠溶液淬灭, 乙酸乙酯(100 mL×3)萃取. 合并有机相, 经无水MgSO4干燥后过滤, 减压浓缩. 粗产物通过硅胶柱层析纯化[V(石油醚)∶V(乙酸乙酯)=20∶1~1∶1梯度洗脱], 获得目标产物3a (31.6 mg), 分离产率达90%.
(Cheng, F.)