



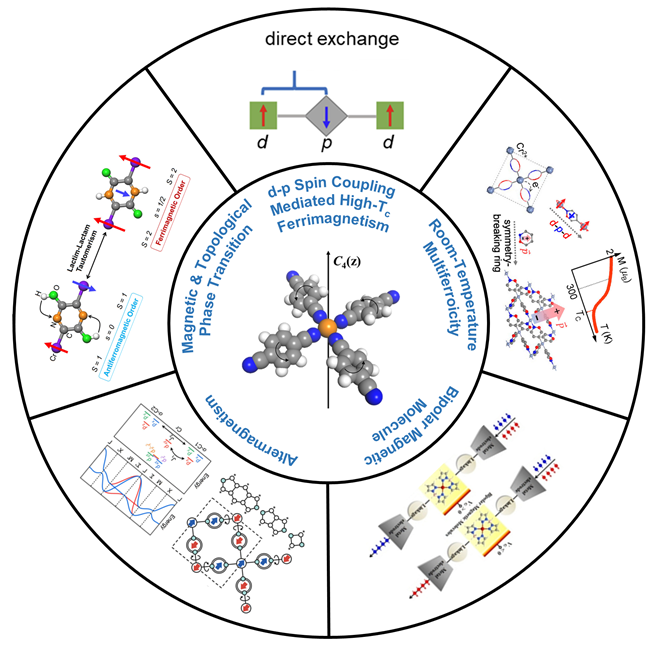
1 引言
2 低维磁性金属有机体系理论设计
2.1 d-p直接交换相互作用
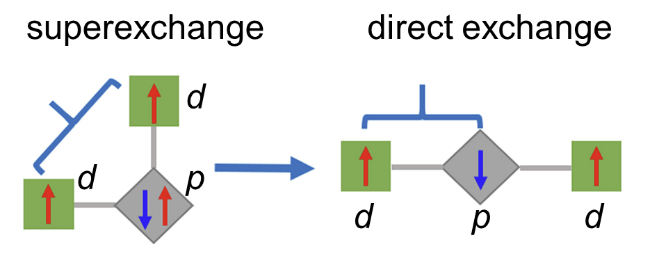

图2 (a)基于反芳香环的金属有机框架实现高居里温度的二维磁性半导体[16]. (b)可剥离的Cr(pyrazine)2单层材料实现高居里温度及电控自旋极化的二维双极磁性半导体[17]Figure 2 (a) Realizing two-dimensional magnetic semiconductors with enhanced curie temperature by antiaromatic ring based organometallic frameworks[16]. (b) Two-dimensional bipolar magnetic semiconductors with high Curie-temperature and electrically controllable spin polarization realized in exfoliated Cr(pyrazine)2 monolayers[17] |
2.2 轨道杂化增强磁耦合

图3 (a)轨道杂化增强提高二维Cr(II)芳香杂环金属-有机框架磁体的居里温度[21]. (b)前沿分子轨道调控二维金属-有机框架磁基态和磁耦合作用[22]Figure 3 (a) Enhanced Curie temperature of two-dimensional Cr(II) aromatic heterocyclic metal-organic framework magnets via strengthened orbital hybridization[21]. (b) Enhancing magnetic ordering in two-dimensional metal-organic frameworks via frontier molecular orbital engineering[22] |
2.3 化学调控磁相变

2.4 二维室温多铁金属有机体系

图5 (a)二维有机-无机室温多铁性材料[26]. (b)二维五元杂环铬(II)金属有机框架中通过配体自旋与晶格对称性调控实现的五重功能集成[27]. (c)二维室温金属有机多铁材料的构筑路径: d-p自旋耦合与结构反演对称性破缺的协同效应[28]Figure 5 (a) Two-Dimensional organic-inorganic room-temperature multiferroics[26]. (b) Quintuple function integration in two-dimensional Cr(II) five-membered heterocyclic metal organic frameworks via tuning ligand spin and lattice symmetry[27]. (c) A route to two-dimensional room-temperature organometallic multiferroics: the marriage of d-p spin coupling and structural inversion symmetry breaking[28] |
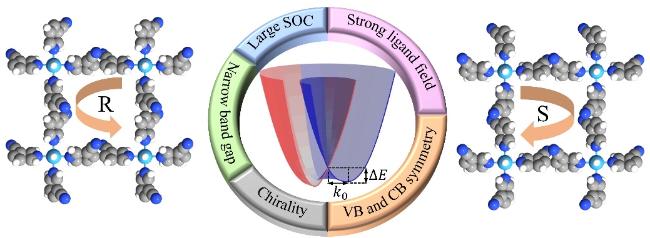
2.5 具有显著Rashba效应的金属有机体系
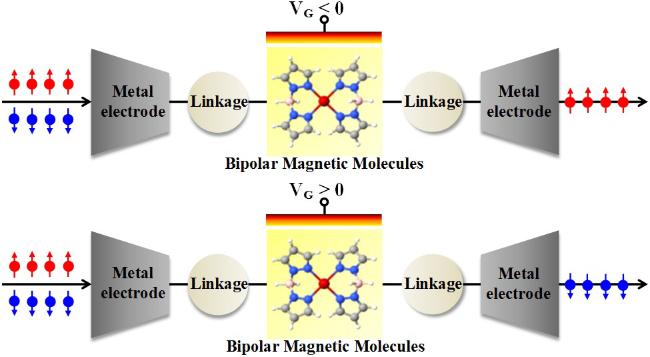
2.6 双极磁性分子
2.7 具有交变磁性的金属有机体系
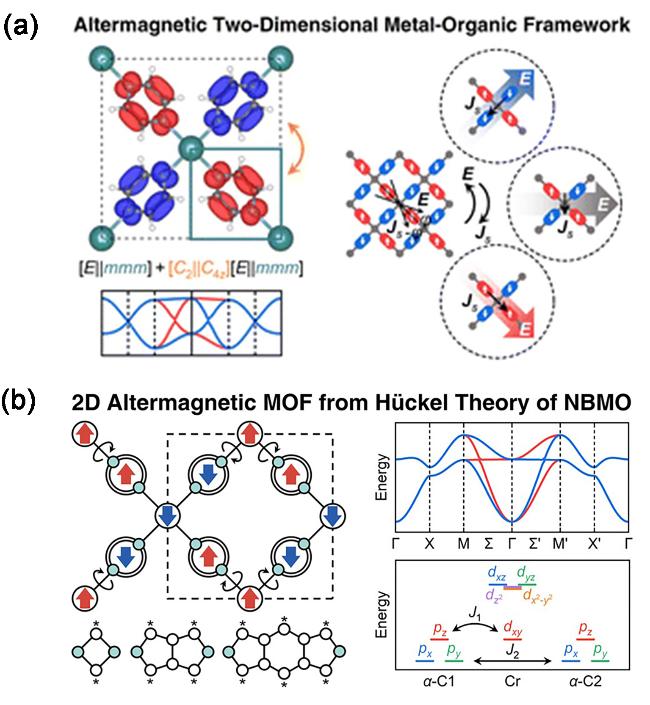
图8 (a)具备电场调控各向异性自旋流功能的二维金属-有机交变磁体[30]. (b)基于非键分子轨道休克尔理论的交变磁性二维金属-有机框架单层材料的逆向设计[31]Figure 8 (a) Realizing altermagnetism in two-dimensional metal-organic framework semiconductors with electric-field-controlled anisotropic spin current[30]. (b) Inverse design of 2D altermagnetic metal-organic framework monolayers from Hückel theory of nonbonding molecular orbitals[31] |
2.8 数据驱动设计“硬”磁性金属有机体系

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图9 (a)以磁各向异性最大化为目标标定钴化学空间的高通量计算研究[33]. (b)过渡金属互连神经网络: 高磁各向异性二维金属-有机框架的机器学习研究[34]Figure 9 (a) Charting regions of cobalt’s chemical space with maximally large magnetic anisotropy: a computational high-throughput study[33]. (b) Transition-metal interlink neural network: machine learning of 2D metal-organic frameworks with high magnetic anisotropy[34] |