







1 引言
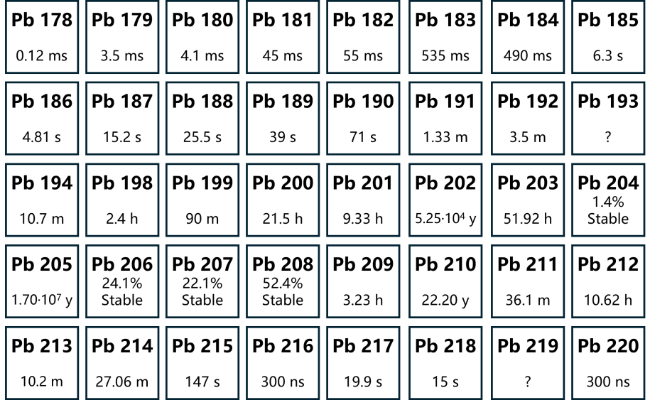
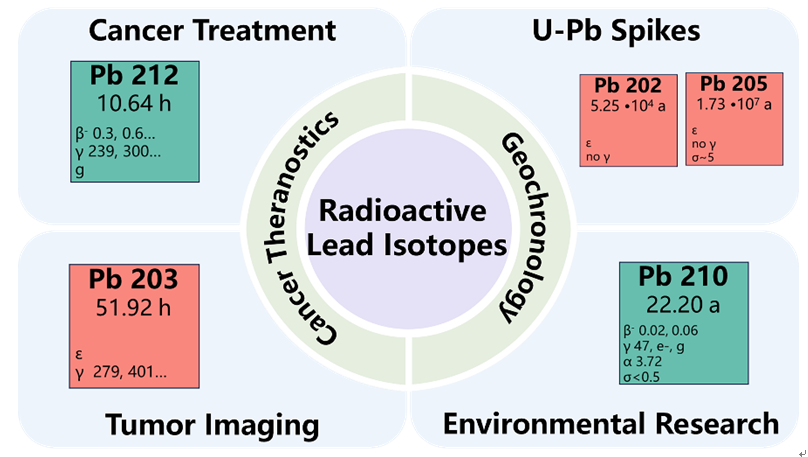
表1 本综述中主要介绍的放射性铅同位素Table 1 Lead radioisotopes mainly discussed in this review |
| 放射性铅同位素 | 半衰期 | 衰变模式 | 应用领域 |
|---|---|---|---|
| 202Pb | 5.25×104年 | EC | 地质年代学研究 |
| 203Pb | 51.9小时 | EC | 核医学癌症诊断 |
| 205Pb | 1.7×107年 | EC | 地质年代学研究 |
| 210Pb | 22.2年 | β- | 环境科学与地球化学研究 |
| 212Pb | 10.6小时 | β- | 核医学癌症治疗 |
2 放射性铅同位素在核医学中的应用
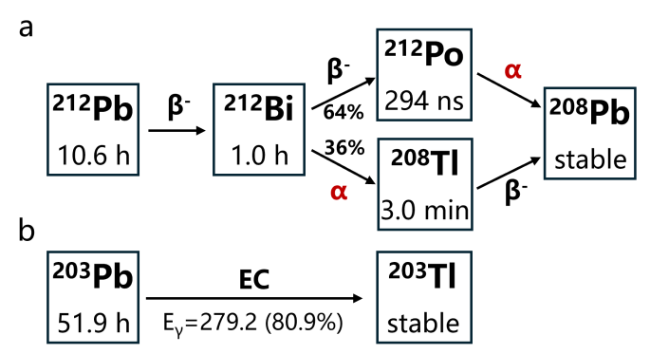
2.1 212Pb的制备及纯化方法
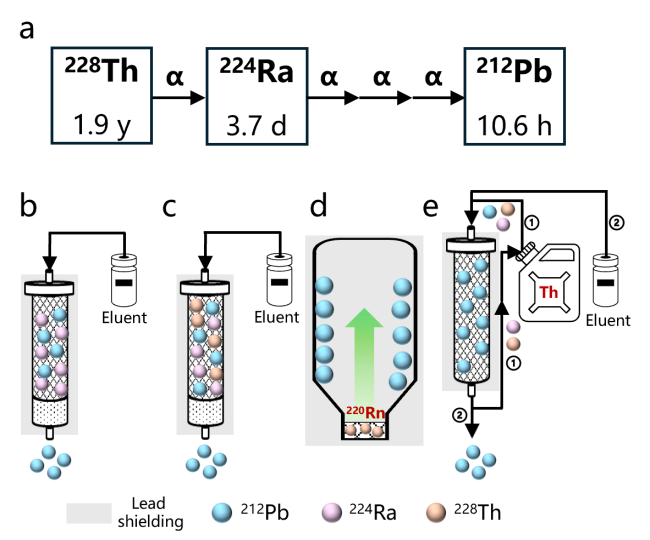
图3 212Pb的制备方法: (a) 228Th可通过α衰变产生224Ra, 进而产生212Pb; (b) 224Ra/212Pb发生器; (c) 228Th/212Pb发生器; (d) 228Th/220Rn/212Pb气相发生器; (e)直接从232Th中分离212PbFigure 3 The production approaches for 212Pb: (a) The decay scheme of 228Th to 212Pb. The daughter radionuclides of 224Ra are not shown; (b) 224Ra/212Pb generator; (c) 228Th/212Pb generator; (d) 228Th/220Rn/212Pb generator; (e) direct separation of 212Pb from natural 232Th |
2.1.1 224Ra/212Pb发生器
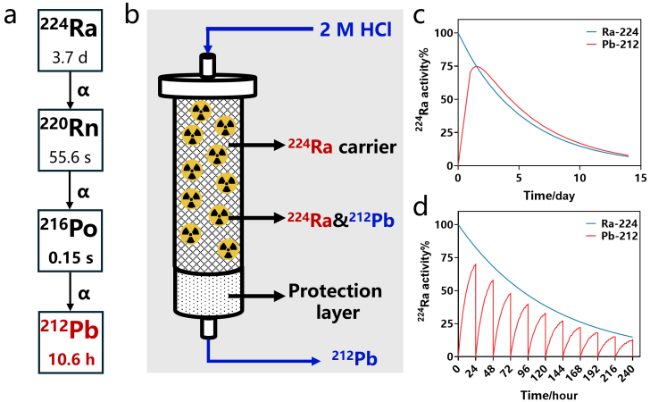
图4 224Ra/212Pb发生器. (a) 224Ra的衰变链; (b)常见224Ra/212Pb发生器组成示意图及淋洗方法; (c) 224Ra/212Pb的放射性平衡曲线; (d) 224Ra/212Pb发生器在10天尺度内的淋洗曲线(假定淋洗效率为100%)Figure 4 The 224Ra/212Pb generator. (a) The decay scheme of 224Ra; (b) Schematic illustration of a 224Ra/212Pb generator including the elution method; (c) The radioactive equilibrium curve between 224Ra and 212Pb; (d) Elution profile of a 224Ra/212Pb generator over ten days assuming 100% elution efficiency |
2.1.2 228Th/212Pb发生器
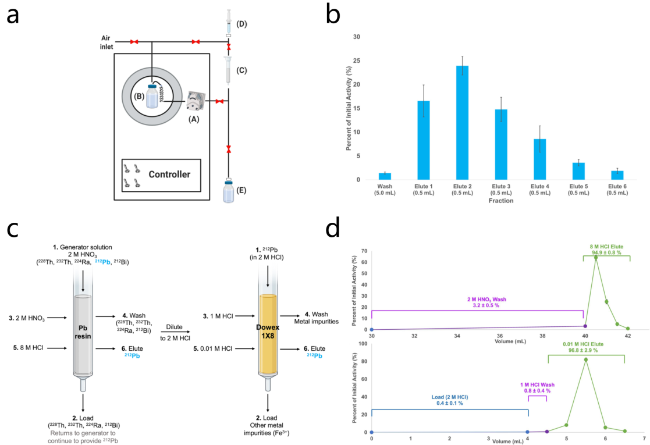
图5 228Th/212Pb的分离. (a) 228Th/212Pb分离装置示意图; (b)基于此装置进行228Th/212Pb分离过程中212Pb的淋洗曲线; (c)改进后的两步228Th/212Pb分离流程示意图; (d)基于改进后流程的212Pb淋洗曲线[41-42]Figure 5 The separation of 212Pb from 228Th. (a) Schematic illustration of the 228Th/212Pb separation set-up; (b) typical 212Pb elution profile using this set-up; (c) scheme of the improved two-step 228Th/212Pb separation procedure; (d) elution profile of 212Pb using the improved procedure[41-42] |
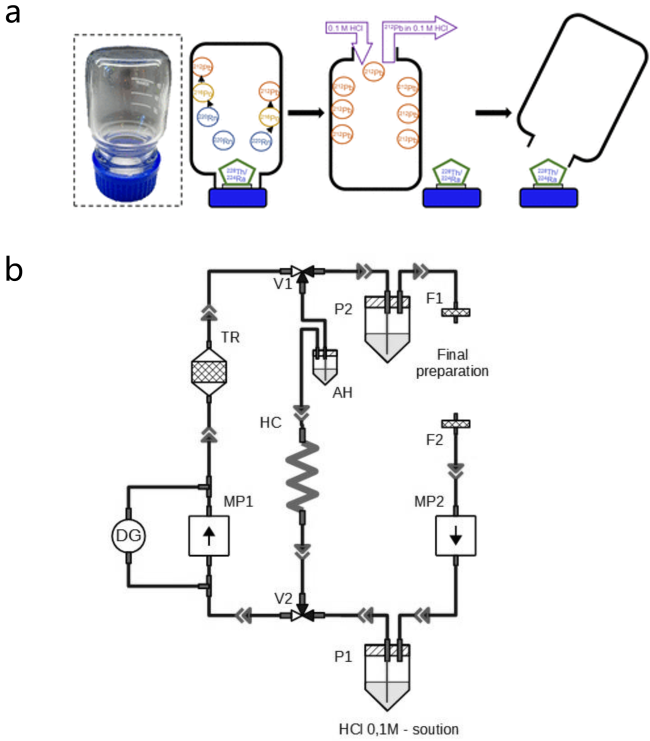
2.1.3 从天然232Th中直接分离212Pb
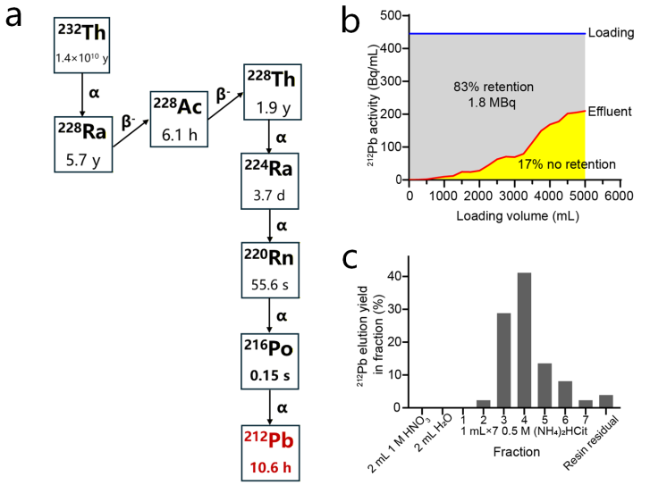
图7 232Th中直接提取212Pb. (a) 232Th的衰变链; (b) 5 L硝酸钍溶液中212Pb在铅特异性树脂上的吸附情况; (c)铅特异性树脂吸附的212Pb的淋洗曲线[48]Figure 7 Direct extraction of 212Pb from 232Th. (a) The decay scheme of 232Th; (b) absorption of 212Pb from 5 L of 232Th solution on the PB resin; (c) elution profile of 212Pb after being absorbed on the PB resin[48] |
2.2 203Pb的制备及纯化方法
2.2.1 用于203Pb制备的核反应
2.2.2 205Tl靶件的制备

表2 文献报道中205Tl电镀液的组成Table 2 Composition of the electroplating buffer for preparing 205Tl targets |
| 靶片材料 | 电镀液组成 | 参考文献 |
|---|---|---|
| 金片 | 1 g [natTl]Tl2O3, 0.5 mol/L EDTA, 0.4 mol/L NaOH, 300 µL 20% BRIJ-35, 1.9 mL 35%水合肼, 33.1 mL超纯水 | [52] |
| 金片或铜片 | 250 mg [natTl]Tl2O3或[205Tl]Tl2O3, 300 μL水合肼, 1 g NaOH, 1.5 g EDTA, 10 mL超纯水 | [55] |
| 银片 | 21 g EDTA, 5 g NaOH, 2.53 mL水合肼, 250 µL BRIJ-35, 90 mL去离子水, 溶解后加入额外2.35 mL 水合肼与250 µL BRIJ-35, 混匀后加至8.475 g [natTl]Tl2SO4或8.949 g of [natTl]TlNO3中, 待固体完全溶解后加入额外250 µL水合肼 | [42] |
2.2.3 203Pb产物的分离纯化方法
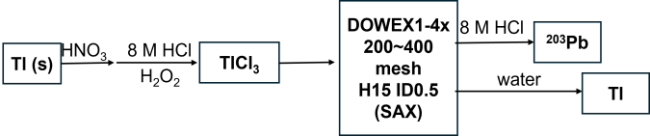
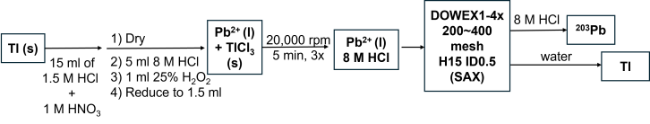
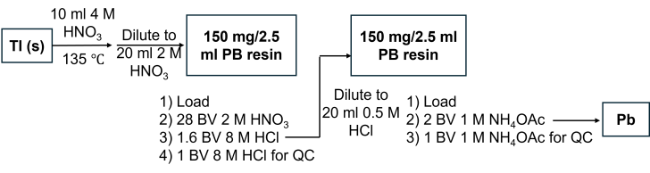
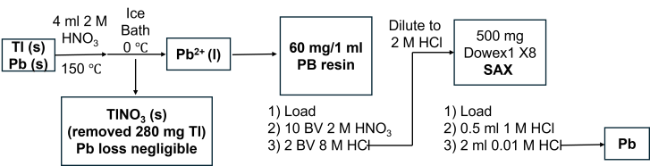
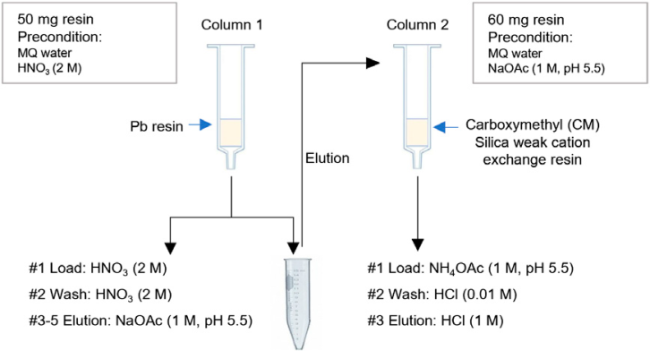
2.3 203Pb/212Pb诊疗一体靶向核药物的临床研究进展
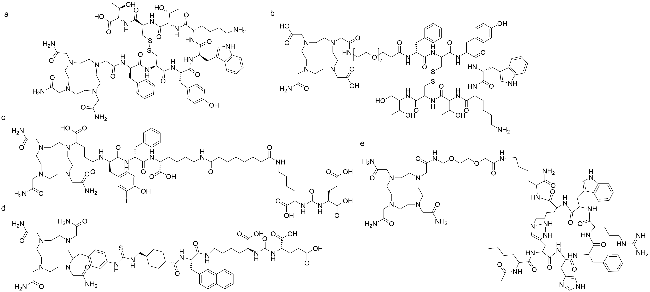
图14 临床研究阶段203Pb/212Pb靶向核药前体分子的化学结构式. (a) DOTAMTATE[38]; (b) VMT-α-NET[75]; (c) ADVC001[76]; (d) NG001[77]; (e) VMT-01[78]Figure 14 Chemical structure of the precursors of the 203Pb/212Pb radiopharmaceuticals under clinical evaluation. (a) DOTAMTATE[38]; (b) VMT-α-NET[75]; (c) ADVC001[76]; (d) NG001[77]; (e) VMT-01[78] |
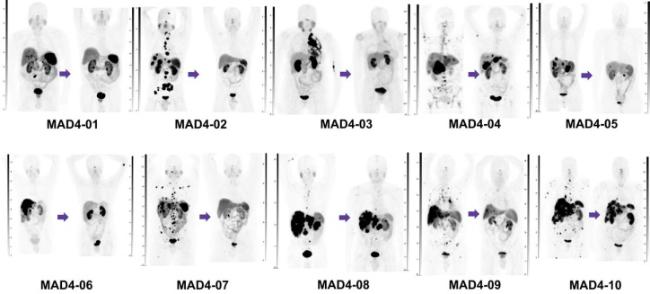
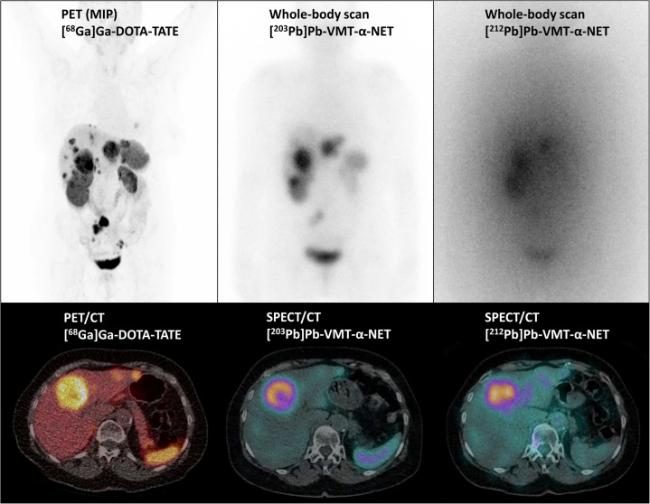
图16 同一神经内分泌肿瘤患者的[68Ga]Ga-DOTATATE PET/CT (左栏)和243 MBq [203Pb]Pb-VMT-α-NET(中间栏)和86 MBq [212Pb]Pb-VMT-α-NET (右栏)静脉注射2.5 h后的SPECT/CT全身成像及横截面成像图[75]Figure 16 The whole body scan and transversal slice image of [68Ga]Ga-DOTATATE PET/CT (left), 243 MBq [203Pb]Pb-VMT-α-NET (middle) and 86 MBq [212Pb]Pb-VMT-α-NET (right) 2.5 h post injection in the same patient[75] |
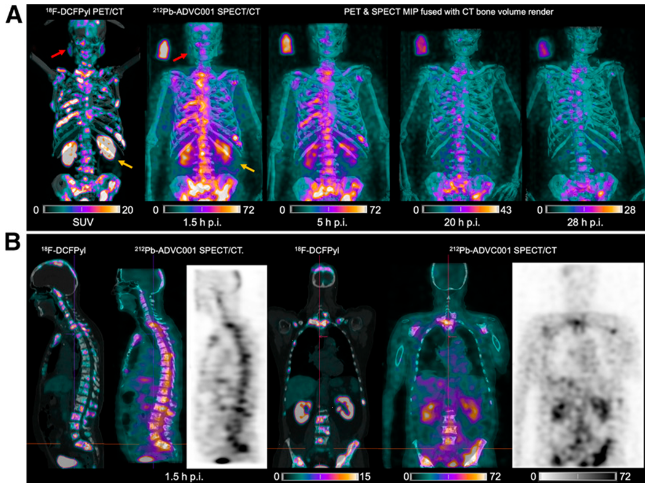
图17 首例[212Pb]Pb-ADVC001人体SPECT/CT成像图. 上图对比了18F-DCFPyl PET/CT与60 MBq [212Pb]Pb-ADVC001 SPECT/CT的成像结果, [212Pb]Pb-ADVC001展现出了快速的肾脏清除和较低的唾液腺摄取. 下图为注射1.5 h后的矢状面和冠状面图像[76]Figure 17 The first SPECT/CT image of [212Pb]Pb-ADVC001 in human. The image above showed the rapid kidney clearance and low salivary gland uptake of [212Pb]Pb-ADVC001, with 18F-DCFPyl PET/CT scan for comparison. The image below shows the sagittal and coronal images 1.5 h post injection[76] |
3 人工铅同位素在地质年代学中的应用
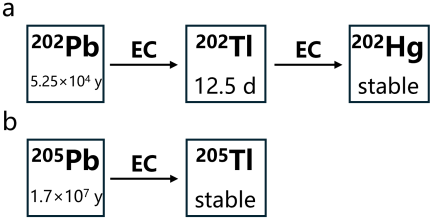
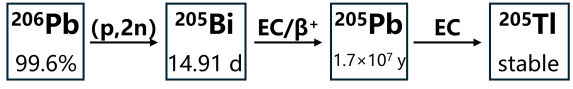
3.1 202Pb和205Pb的衰变性质
3.2 202Pb的制备与纯化方法
3.3 205Pb的制备与纯化方法
4 放射性铅同位素在环境科学中的应用
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 总结与展望
表4 本综述涉及的放射性铅同位素的主要来源汇总Table 4 A summary of the sources the lead radioisotopes discussed in this review |
| 放射性铅同位素 | 制备设施/方法 | 核反应/衰变链 |
|---|---|---|
| 202Pb | 回旋加速器 | 203Tl(p,2n)202Pb |
| 203Pb | 回旋加速器 | 203Tl(p,n)203Pb或205Tl(p,3n)203Pb |
| 205Pb | 回旋加速器 | 206Pb(p,2n)205Bi(EC)205Pb |
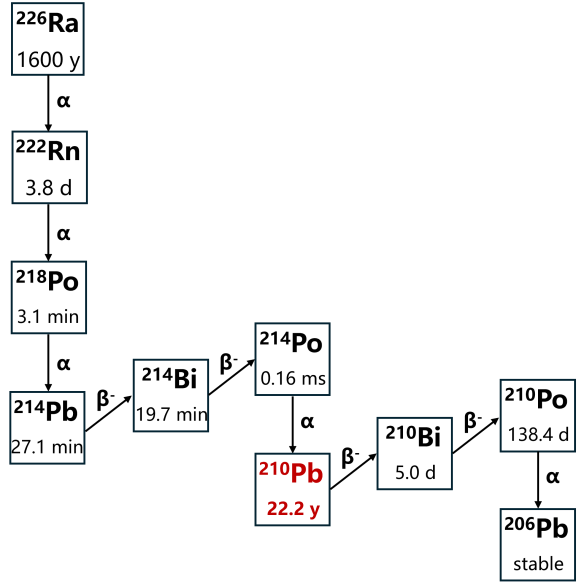
| 210Pb | 226Ra衰变生成 | 226Ra(α)222Rn(α)218Po(α)214Pb(β-)214Bi(β-)214Po(α)210Pb |
| 212Pb | 核反应堆 | 226Ra(n,γ)227Ra(β-)227Ac(n,γ)228Ac(β-)228Th(α)224Ra(α)220Rn(α)216Po(α)212Pb |
| 核废料分离 | 232U(α)228Th(α)224Ra(α)220Rn(α)216Po(α)212Pb | |
| 天然钍分离 | 232Th(α)228Ra(β-)228Ac(β-)228Th(α)224Ra(α)220Rn(α)216Po(α)212Pb |