


1 引言
1847年, 意大利化学家阿斯坎诺•索布列罗首先合成了硝化甘油(Nitroglycerin, NG)[1-2]. 实际生产的硝化甘油安定性较差, 易因震动或撞击而爆炸. 瑞典化学家阿尔弗雷德•诺贝尔将硅藻土和硝化甘油混合, 发明了“代那买特”炸药, 显著提高了硝化甘油的安全性[3-4]. 如今, 硝化甘油不仅是双基推进剂、复合推进剂、枪炮发射药和工业炸药的重要组分, 也是医学领域的救命 药[5-6]. 然而, 工业品硝化甘油具有缓慢自分解的倾向, 且会逐渐加速, 不仅会变质, 还会带来巨大的安全隐患. 这种分解使它的化学、物理性质发生显著变化[7]. 在火工品生产发生的重大事故中, 硝化甘油爆炸事故占1/4以上, 部分统计数据显示, 在630次事故中有158次涉及硝化甘油[7-10]. 即便已经经历了170多年的漫长岁月, 应用已如此广泛, 但是工业硝化甘油产品与生俱来的安全问题仍未解决, 时刻威胁着生产、存储、运输和应用环节的安全.
硝化甘油是双基推进剂的主要成分之一, 也是复合推进剂中常用的高能增塑剂[5-6]. 对推进剂的能量和力学性能都有较大影响. 文献指出, 在储存期间, 硝化甘油会释放出氮氧化物, 从而加速推进剂的老化[11-12]. 由于对硝化甘油的分解机理不够了解, 在很大程度上限制了它的使用和贮存[13]. 与硝化甘油有关的热分解实 验[14-18]和理论研究工作已有一些报道. 实验测得的初始热分解能垒为41.3~48.0 kcal/mol[18]. 理论计算[19-27]的初始分解能垒分布范围从4.8 kcal/mol[22]到69.6 kcal/ mol[25]. 这可能是由于计算方法或模型处理的差异造成的. 可见, 依据这些研究结果, 理论上很难形成对机理的一致观点.
硝化甘油的相关问题充满了神秘感和不确定性. 因此, 首先要理清思路, 澄清一些基本认知. 我们认为, 硝化甘油分子具有原生的、内在的及固有的热稳定性.以下的事实支持这一观点: (1)硝化甘油的发明者阿斯坎诺•索布列罗合成的硝化甘油存放了70年后进行检验, 并没有显著分解. 这说明硝化甘油经过适当处理, 能够稳定长期存储. 也意味着硝化甘油自身具有较好的热稳定性. (2)实验表明, 硝化甘油分解峰温度达206 ℃(5 MPa), 表明硝化甘油的分子结构是相对“牢固”的[28-29]. (3)一般认为初始分解的引发键是硝酸酯键O—NO2的均裂, 该键的键能为(49.3±0.3) kcal/mol[30], 均裂反应能垒应该接近这一数值, 并不算非常低. 因此, 我们有理由推测, 常温下硝化甘油的缓慢热分解, 可能主要是其他条件影响造成的.
曲开社[33]用统计方法研究了原料甘油中的杂质对硝化甘油产品安定性的影响程度, 利用分项评分得到如下结果: 对硝化甘油产品安定性有显著影响的杂质依次为丙烯醛、糖类、Zn2+、Mg2+、Cu2+; 有一定影响的杂质依次为甲醛、Fe2+、乳酸、Cl-、水、F-和甘油醛; 有影响的杂质依次为1,2-丙二醇、丙酮、乙酸和 . 我们认为, 甘油中的丙烯醛、糖类及甲醛等还原性有机物不太可能在浓硝酸/浓硫酸的硝化条件下不被破坏而继续存留在硝化甘油产品中. 因此, 对硝化甘油的研究可不考虑这些杂质. Aburawi等[34]研究了HCl和NaOH对硝化甘油安定性的影响, 发现碱的影响更大. 硝化甘油体系中同时存在NO、NO2和H2O时, 可以快速分解. 当分解产物含有H2O和HNO3时, 分解的加速十分剧烈. 此外, 光照对硝化甘油的安定性也有很大影响[31-32].
本文的研究目的是阐明硝化甘油常温下缓慢分解的真实机理. 通过系统计算“无杂质”和“含杂质”条件下的分解机理, 揭示缓慢分解的真实历程和影响因素. 首先, 对于广泛报道的单分子分解机理进行了梳理和重新计算分析. 然后, 我们挑选对硝化甘油稳定性影响较明显的杂质进行了研究, 包括H2O、残酸(HNO3)、Mg2+、Zn2+, OH-、 和Cl- .
2 结果与讨论
2.1 硝化甘油传统单分子热分解机理的重新讨论
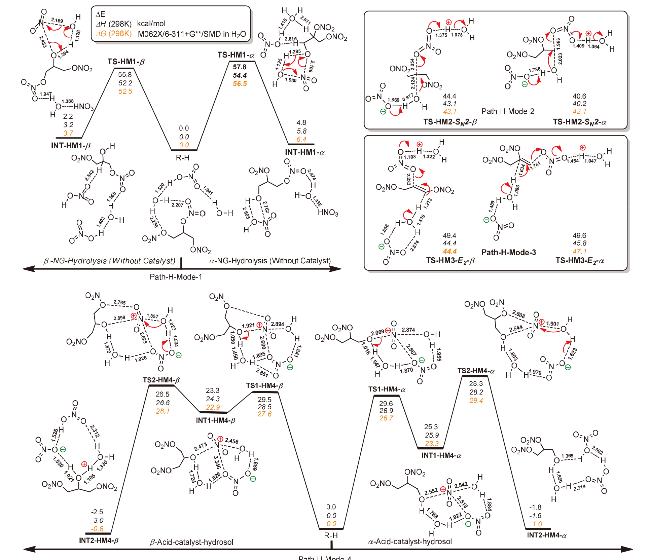
图1总结了文献中的硝化甘油单分子分解机理. 反应模式Mode-1是硝酸酯中O—NO2键的均裂机理, 该机理生成•NO2自由基; 与Mode-1接续, Mode-2中, •NO2自由基的O原子拔H生成HONO, 并促使硝化甘油继续分解. 目前, 很多研究者对Mode-2比较认可. 在此基础上, 我们进一步提出了Mode-2-2和Mode-2-3两条新路径, 分别是•NO2自由基的N原子拔氢, 以及•NO2自由基的N原子攫取硝酸酯的•NO2, 生成N2O4和烷氧基自由基中间体的过程. Mode-3是传统的分子内消除HONO的机理[21,24,28-29]. 我们对硝化甘油单分子初始分解的这些途径进行了重新研究, 对硝化甘油分子在气相和溶剂化条件下进行了构象搜索[35]. 正文中只给出了气相的计算结果.
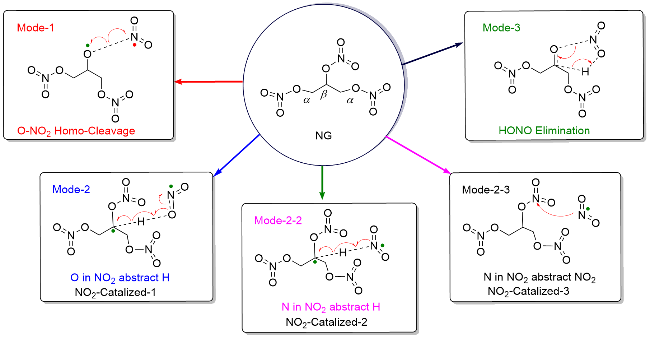
2.1.1 O—NO2键均裂机理(Path-Mode-1)
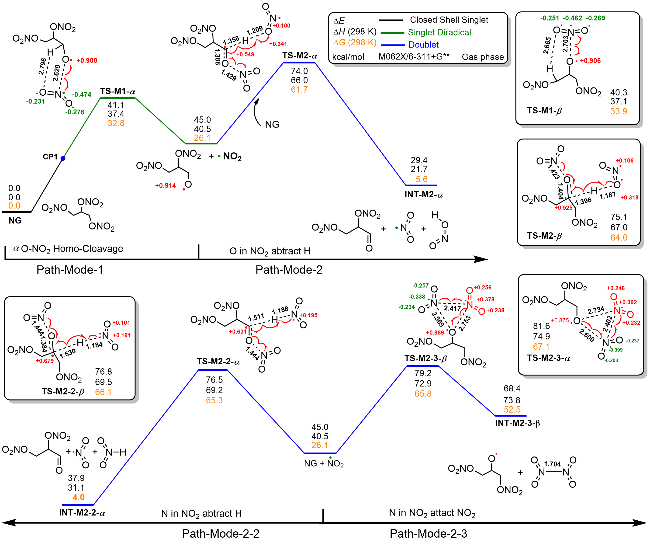
2.1.2 •NO2催化机理(Mode-2, Mode-2-2和Mode-2-3)
在硝化甘油的单分子初始热分解路径中, Mode-2被研究者认为是主要分解路径, 即中间产物•NO2的O原子拔硝化甘油分子C—H键的H, 并引发进一步的分解. 如图2所示, 在反应路径Path-Mode-2中, 通过过渡态TS-M2-α (61.7 kcal/mol), •NO2拔取C—H键的H原子, 生成HONO, 同时C原子上的自由基转移到与其相连的硝酸酯基团上, 硝酸酯基团脱去一个•NO2自由基生成羰基化合物INT-M2-α. 由于此时体系中的•NO2是硝化甘油分解的产物, 键均裂生成•NO2后体系能量升高了26.1 kcal/mol. 这样, 在初始分解时, •NO2催化步骤的总体能垒高达61.7 kcal/mol. 因此, 除非•NO2是外界引入或是通过更容易的途径产生, 或者经过长时间积累达到相当大的浓度, Path-Mode-2在常温下的初始分解步骤是不太可能实际发生的. 以往人们认为•NO2攫氢催化硝化甘油分解是比较合理的机理, 是因为有些理论研究单独讨论•NO2促进的反应机理, 未考虑•NO2的生成步骤. 即他们只考虑了单步反应能垒, 而不是总体反应能垒. 这样一来, 就把“NG+•NO2”看成了反应的能量零点, 没有考虑总体反应路径, 即硝化甘油才是最低的能量参考点. 把•NO2促进的机理单独列出, 从而没有认识到总体能垒较高. •NO2是热力学不稳定的产物, 累积到高浓度的可能性比较小. 因此, 我们推测, 分解反应加速的原因, 可能是•NO2转化而成的某种热力学较为稳定的分子. Mode-2单一步骤的能垒为35.6 kcal/mol, 比O—NO2键均裂路径的能垒(32.8 kcal/mol)高2.8 kcal/ mol. 只有当体系中•NO2(或热力学更稳定的等价物)的浓度积累达到一定程度, 即体系经过“诱导期”后, 这种反应机理才是可能的.
此外, 我们还计算了两种新机理Mode-2-2和Mode-2-3. 在反应模式Mode-2-2中, •NO2的N原子拔H; 在反应模式Mode-2-3中则是•NO2从硝酸酯基拔取•NO2自由基. 计算结果表明, 这两种机理并没有特别的优势.
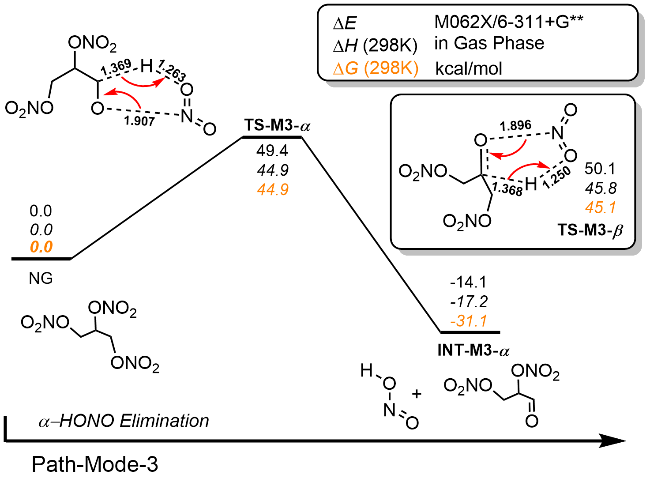
2.1.3 分子内消除HONO机理(Path-Mode-3)
如图3所示, C原子上硝酸酯基团O—NO2中的NO2和C—H键的H原子通过五元环协同过渡态TS-M3-α消除一分子HONO生成醛, 能垒为44.9 kcal/mol. 这一能垒比其它反应模式能垒高. TS-M3-β能垒为45.1 kcal/mol.
2.1.4 溶剂化效应
考虑硝化甘油自身作溶剂时, 我们依据手册[37]中硝化甘油的参数进行计算. 介电常数19.25 D(德拜), 表面张力49.48 mN•m-1, 折射率为1.4786.
表1中的结果表明, 溶剂化对硝化甘油反应路径能垒影响较小. 这是因为这些反应中, 反应物、过渡态的极性较小, 溶剂化效应不明显. 水作溶剂时, Path-Mode- 2能垒下降稍明显.
表1 硝化甘油单分子分解各反应路径的能垒[ΔG/(kcal•mol-1)]Table 1 Energy barriers of nitroglycerine unimolecular decomposition [ΔG/(kcal•mol-1)] |
| Pathway | Gas phase | In NG | In H2O | |||||
|---|---|---|---|---|---|---|---|---|
| α | β | α | β | α | β | |||
| Path-Mode-1 | 32.8 | 33.9 | 35.9 | 35.5 | 35.0 | 33.9 | ||
| Path-Mode-2 | 61.7 | 64.0 | 63.6 | 64.4 | 58.9 | 60.3 | ||
| Path-Mode-2-2 | 65.3 | 66.1 | 62.9 | 64.4 | 61.0 | 62.0 | ||
| Path-Mode-2-3 | 67.1 | 65.8 | 67.2 | 66.2 | 64.9 | 64.4 | ||
| Path-Mode-3 | 44.9 | 45.1 | 47.0 | 47.8 | 45.7 | 46.1 | ||
2.2 硝化甘油的质子酸催化水解机理
在硝化甘油中总是存在少量H2O以及未充分洗净的残酸(HNO3). 硝化时混酸中的硝酸可能会导致有部分NO2和NO残存在硝化甘油中. 而且, 硝化甘油的缓慢分解也会产生酸. 我们对无催化和质子酸催化的硝化甘油水解机理进行了系统研究.
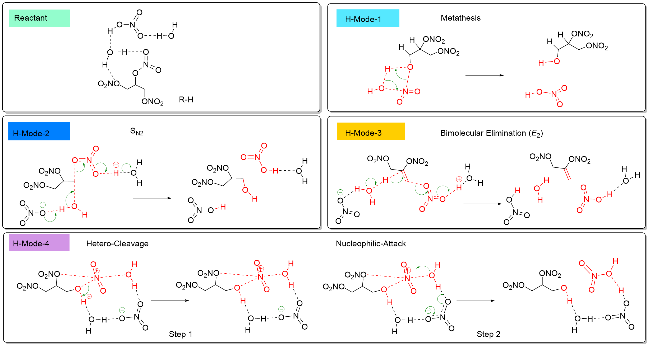
2.2.1 无催化水解反应机理
作为参照, 我们首先计算了没有酸催化时的水解机理. 即只考虑体系中存在水时的水解机理(H-Mode-1, 保持其中HNO3不参与催化作用). 此时, 水分子直接和硝酸酯基团反应: 水分子的氧原子进攻硝酸酯基团的N原子, 水分子的OH和NO2结合; 而水分子的另外一个H原子和硝酸酯基团C—O的O原子结合. 这种机理在形式上类似于复分解反应. 该机理的能垒达到52.5 kcal/mol. 我们预期五元环过渡态会减小张力而能量更优, 但是计算结果表明, 五元环过渡态能量并不会降 低[35]. 显然, 该机理能垒太高, 室温下是不可能发生反应的, 这意味着硝化甘油对纯水是稳定的. 这和硝化甘油通常使用水封保存的事实相一致. 这与文献中的记载也一致, 意大利保存的阿斯坎诺•索布列罗(Ascanio Sobrero)实验制得的硝化甘油, 历经了70年仍未见明显分解的迹象[1,31]. 这表明硝化甘油在足够纯净的情况下, 具有较好的安定性, 可以长期保存. 计算结果和实验相符. 为了避免从原料中带入杂质, 美、日、英及法等国对硝化甘油生产中甘油的质量要求极高, 其纯度标准通常需达到99%以上, 有些甚至规定为99.8%[31].
2.2.2 质子酸催化的硝化甘油水解机理
H-Mode-2反应模式中, 硝酸酯基团在被质子活化的情况下, 被水分子通过SN2机理取代, 生成硝酸和甘油二硝酸酯中间体. 经过这一途径, 最初的一分子催化剂硝酸保持不变, 并且又生成了一分子新的硝酸. 该路径是一个协同过程: 羟基氧原子的孤对电子进攻硝酸酯基上的碳原子, 同时硝酸酯基从碳原子上离去并与质子结合, 生成一分子硝酸. 在该过程中, 会通过氢键稳定H2O分子中的氢, 增强H2O分子进攻碳原子的能力, 并在反应后接受H2O分子释放的质子. 然而, 碱性较弱, 该路径的最低能垒为42.1 kcal/mol, 虽然比H-Mode-1显著降低, 但明显高于O—NO2键均裂的能垒. 因此, 该机理可以被排除.
在H-Mode-3中, 硝酸酯在质子活化下, 发生双分子消除反应(E2), 消去硝酸酯基团及其β-H生成烯烃衍生物和一分子硝酸. 该机理也是一个协同过程. 其中硝酸根离子通过水分子夺取质子, 同时硝酸酯基团作为 负离子离去, 生成一分子硝酸. 然而, 该反应路径的能垒也比较高(44.4 kcal/mol), 因此也可以排除.
H-Mode-4水解机理分为两步. 第一步(Step 1), 在质子活化条件下, 硝酸酯基团的O—NO2键发生异裂, 释放出硝酰阳离子 . 第二步(Step 2), 硝酰阳离子 进一步和水分子反应生成硝酸. 如图5中TS2-HM4-β所示, 阴离子通过形成氢键活化水分子, 使水分子中O原子的孤对电子更容易进攻硝酰阳离子带正电荷的N原子, 生成一分子HNO3. 我们认为, 这一过程恰是硝化反应的逆反应. 现在比较公认的甘油硝化机理, 是HNO3首先被硫酸脱水, 生成硝酰阳离子 , 进而硝酰阳离子 和醇羟基发生反应生成酯. 值得注意的是, 初始的一分子HNO3在水解反应后仍然存在, 而且每次反应都新生成一分子硝酸. 这样, 反应在经过诱导期后会显著加速.
由于过渡态有许多可能的构象, 找到能量最低的构象十分重要. TS2-HM4-β是酸催化水解路径的决速步骤, 如能进一步优化是很有意义的. 我们仔细分析了TS2-HM4-β的结构(图6), 发现硝酸酯基团的O(1)带有负电荷-0.391, 离去的硝酰阳离子的O(2)带有负电荷-0.095, O(5)带有负电荷-0.821. 然而, O(1)、O(2)的距离却只有2.765 Å, O(1)、O(5)的距离也只有2.784 Å. 从常识判断, 他们之间应该存在较大的排斥力. 因此, 如果把这个硝酸酯基团围绕C—C键旋转60°(图6中红色箭头), 使O(1)远离O(2)和O(5), 减小排斥力, 且同时H(6)靠近O(2)和O(5), 形成分子内氢键, 进一步降低体系能量. 按照这样的设想, 我们优化了过渡态TS2-HM4-β2. 但让人吃惊的是, 这个新过渡态的能量居然上升了, 能垒升高至29.1 kcal/mol. 这意味着, 进行结构分析时, TS2-HM4-β中的某种稳定作用被忽略了. 于是, 我们分析了TS2-HM4-β的分子轨道和 结构片段的分子轨道. 结果发现, O(1)和O(2)有明显的同相轨道重叠. O(2)和O(5)之间也有类似作用. 缺电子的 结构的π轨道和O(4)也有轨道重叠. 因此, 我们认为, 是缺电子的 结构片段和富电子结构片段之间的轨道作用改变了简单静电作用的总体结果. 这一结构在某种程度上可以理解为非金属阳离子形成的络合物.
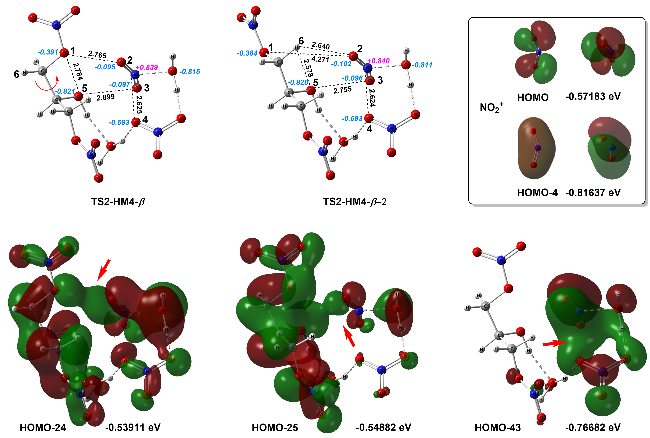
表2 硝化甘油各水解反应路径的活化能[ΔG/(kcal•mol-1)]Table 2 Energy barriers of hydrolysis reaction pathways of nitroglycerin [ΔG/(kcal•mol-1)] |
| Pathway | SMD in NG | SMD in H2O | |||
|---|---|---|---|---|---|
| α | β | α | β | ||
| Path-H-Mode-1 | 51.9 | 54.2 | 56.5 | 52.5 | |
| Path-H-Mode-2 | 49.3 | 50.9 | 42.1 | 43.1 | |
| Path-H-Mode-3 | 54.5 | 51.5 | 47.1 | 44.4 | |
| Path-H-Mode-4 | 39.0 | 32.5 | 29.4 | 28.1 | |
2.3 金属络合物参与的硝化甘油分解机理
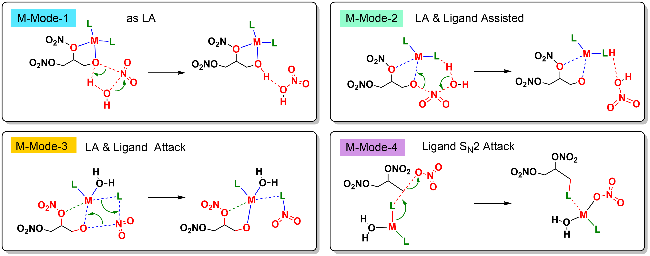
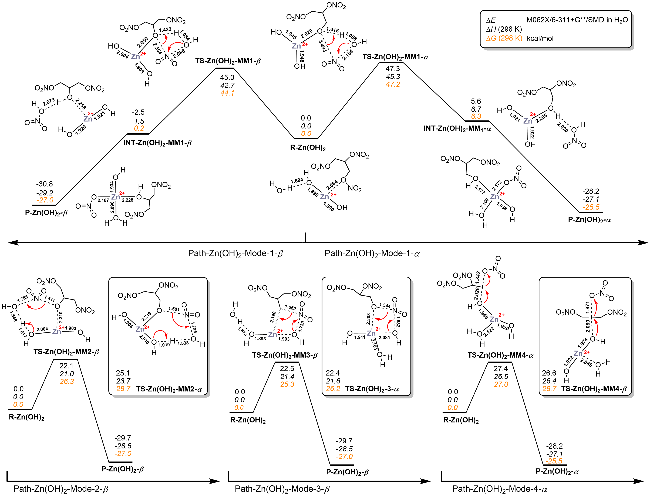
图7 金属络合物促进的硝化甘油水解机理Figure 7 Mechanism of metal complex promoted nitroglycerine hydrolysis |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图8 金属络合物促进的硝化甘油水解机理(键长单位: Å)Figure 8 Mechanism of metal complex promoted nitroglycerine hydrolysis (The bond lengths are in angstroms) |
表3 金属离子催化硝化甘油的路径比较Table 3 Comparison of hydrolysis pathways of nitroglycerin |
| M=Zn2+ | M=Mg2+ | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OH- | Cl- | OH- | Cl- | ||||||||||||||
| α | β | α | β | α | β | α | β | α | β | α | β | ||||||
| M-Mode-1 | 47.2 | 44.1 | 45.0 | 44.5 | 45.9 | 42.1 | 43.5 | 38.6 | |||||||||
| M-Mode-2 | 28.7 | 26.3 | 54.1 | 48.3 | 29.2 | 30.1 | 52.6 | 46.1 | |||||||||
| M-Mode-3 | 26.2 | 25.3 | 21.3 | 22.0 | |||||||||||||
| M-Mode-4 | 27.0 | 27.5 | 36.4 | 39.9 | 27.3 | 26.7 | 43.3 | 47.7 | |||||||||
在反应模式M-Mode-1中, 金属离子与硝酸酯基的O原子配位. 金属离子作为路易斯酸活化O—NO2键, 从而降低水解所需的能垒.
反应模式M-Mode-2中, 除金属离子作为路易斯酸进行活化外, 配体L通过和水分子形成氢键, 活化水分子, 增加氧原子的电子密度, 协助和促进水分子进攻, 并稳定其释放的质子, 使反应更容易, 生成一分子硝酸.
反应模式M-Mode-3是反应模式M-Mode-2的变体. 金属离子作为路易斯酸进行活化, 配体L则直接进攻硝酸酯基团, 特别当配体L是OH-时. M-Mode-3和M-Mode-2中, 都存在中心离子的路易斯酸活化作用和配体的协助或直接进攻作用. 这两种机制协同发挥作用.
反应模式M-Mode-4中, 金属离子不和硝酸酯基团的氧原子配位, 金属离子的配阴离子L从背后以SN2方式进攻和硝酸酯相连的C原子. 硝酸酯以 负离子的形式离去. 这种活化模式只有配体的亲核性起作用, 而中心离子不起活化作用.
图8中M-Mode-1的反应路径Path-Zn(OH)2-MM1- β中, 反应络合物R-Zn(OH)2经过过渡态TS-Zn(OH)2- MM1-β, 生成中间体INT-Zn(OH)2-MM1-β. 该中间体中, 包含一分子新生成的HNO3, 其尚未和Zn(OH)2的OH-中和. 中和之后, 生成P-Zn(OH)2-β并释放能量. 因此, 经过两轮反应, Zn(OH)2的OH-将耗尽, 转化为Zn(NO3)2, 只能作为路易斯酸催化水解反应, 且活性不高. 有意思的是, Zn(OH)2、Mg(OH)2都能很容易地产生INT-Zn(OH)2-MM1-β这类含有硝酸的中间体, 特别是对于M-Mode-2和M-Mode-3, 能垒分别只有25.3和21.3 kcal/mol. 我们知道, 硝化甘油和碱容易反应, 销毁少量的硝化甘油样品正是利用这一反应[2]. Zn(OH)2、Mg(OH)2在作为ppm级浓度的杂质, Zn(OH)₂-MM1-β中这一分子HNO3非常可能扩散到大量的硝化甘油分子中去. 此时, HNO3再和极少量OH-碰撞中和的概率非常小, 而和硝化甘油分子碰撞的概率极大. 因此, 这一分子HNO3就可能成为硝化甘油水解的催化剂. 直到随着水解的进行, HNO3累积到一定浓度, 才会中和掉这些少量的OH-. 而在此之前Zn(OH)2也一直在进行催化水解. 因此, 极少量碱性杂质参与的硝化甘油水解, 虽然表面上看是化学计量反应, 但却可能是诱发酸催化的种子.
根据软硬酸碱理论(HSAB)[38], 硬酸倾向于与硬碱结合, 软酸倾向于与软碱结合. O电负性高(3.44)、孤对电子难以极化, 是硬碱, 倾向于通过强静电作用与硬酸结合. Mg2+是典型的硬酸, 电荷高, 离子半径小(约0.71 Å), 极化性低. 而Zn2+被归类为交界酸或接近软酸, 其离子半径稍大(约0.74 Å), 极化性高于Mg2+. 因此, 和O配位作为路易斯酸对硝酸酯进行活化, Mg2+的反应活性应大于Zn2+. 从表3的反应能垒数据可以看出, 对于单纯的路易斯酸活化模式M-Mode-1, Mg(OH)2的活性略高, 其能垒比相应的Zn(OH)2的都低. Mg(OH)2是典型的中强碱, 而Zn(OH)2的碱性较弱, 甚至表现出酸碱两性. 因此, 路易斯酸性和配体碱性都起直接作用, 而且相互协同的活化模式M-Mode-3是最好的活化模式, 且Mg(OH)2活性高于Zn(OH)2 (21.3 kcal/mol vs. 25.3 kcal/mol). 由于 和Cl-的碱性都很弱, 因此反应能垒高, 反应活性差. 因此, 有些活化模式略去未计算.
3 结论
采用M06-2X/6-311+G**方法系统地研究了硝化甘油的分解机理, 包括单分子热分解、无催化水解、酸性杂质(HNO3)催化水解以及金属盐/碱杂质(Zn2+/Mg2+阳离子与OH⁻/ /Cl⁻阴离子的组合)参与的分解反应.
(1)在单分子分解路径中, O—NO2均裂产生•NO2自由基是最优的初始反应路径, 与很多报道一致.
(2)在酸性杂质存在时, 我们探索了多种反应模式. 最优的反应模式中, 在质子的活化下, 硝酸酯基团的 O—NO2键发生异裂, 释放出硝酰阳离子 ; 硝酰阳离子进一步和水分子反应生成硝酸. 酸催化水解的能垒为28.1 kcal/mol, 比单分子分解机理的O—NO2均裂能垒下降了5.8 kcal/mol, 是更合理的室温缓慢分解机理. 而且, 随着水解不断进行, 释放出越来越多的HNO3, 使分解反应速度不断加快.
(3) Zn(OH)2和中强碱Mg(OH)2与硝化甘油的反应能垒分别为25.3和21.3 kcal/mol. 该反应模式中存在路易斯酸活化和阴离子配体直接进攻硝酸酯的协同作用, 反应速度较快. 碱作为杂质存在时, 含量很少, 虽然该反应是化学计量反应, 但是, 由于反应生成含有硝酸的中间体, 硝酸和硝化甘油碰撞的概率远大于和碱碰撞而中和的概率, 由此可能会引发酸催化分解反应. 而酸催化过程一旦启动, 就会不断自加速. Zn(NO3)2、Mg(NO3)2、ZnCl2和MgCl2催化水解的反应活性不高, 其反应活性主要受阴离子的碱性和阳离子的路易斯酸性强弱的影响.
我们认为, 酸催化机理应该最接近硝化甘油常温下缓慢分解的真实机理. 水解过程中, 硝酸不断积累, 反应速度逐渐加快. 此前的理论研究多集中于硝化甘油的单分子分解路径, 而且没有考虑杂质的影响, 不足以解释常温下的缓慢分解现象. 我们的研究为理解硝化甘油的分解机理提供了新的思路和参考.
4 计算程序和方法
采用Gaussian16程序包[39], 使用对称破缺密度泛函方法, 在M06-2X/6-311+G**计算水平上[40-42], 对硝化甘油分解反应的机理进行系统的研究和梳理. 共价键的均裂在热分解过程中十分普遍. 在核间距平衡位置, 系统处于“闭壳层单重态”. 随着键的拉长, 系统逐渐转变为“开壳层单重态双自由基”[36,43-46]. 使用对称破缺的密度泛函方法(Broken-Symmetry UDFT method)处理共价键均裂过程, 在BS-UM06-2X/6-311+G**水平上对硝化甘油初始分解机理O—NO2均裂进行研究. 对过渡态、中间体及产物的分子结构进行了全优化和振动分析计算, 确认了中间体没有虚频, 过渡态有且只有一个虚频. 对于关键反应过程的过渡态, 进行了内禀反应坐标计算(IRC)[47], 证明过渡态连接着正确的反应物和产物.
(Zhao, C.)