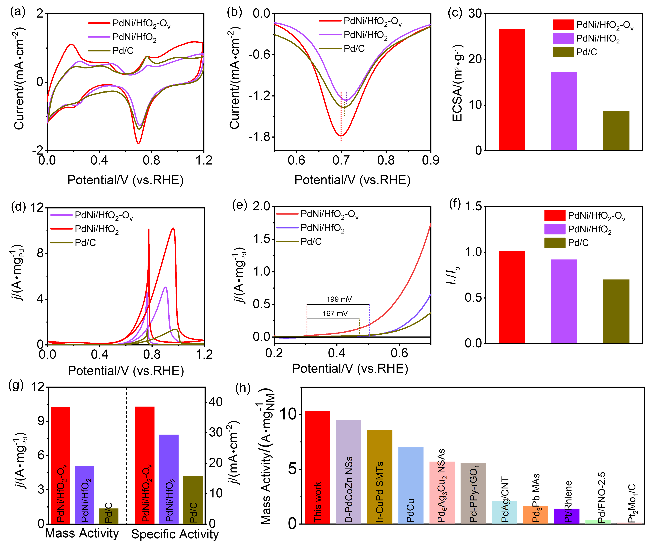
采用循环伏安法在氮气饱和的1 mol/L KOH和1 mol/L KOH+1 mol/L EG溶液中评估了材料的电化学性能(支持信息图S3). 发现材料中不含有Pd时, Ni/HfO
2-O
v和Ni/HfO
2对EGOR均未呈现出明显的反应活性, 表明材料的主要催化活性位点是Pd. 如
图4a所示, 催化剂的活化过程最初是在氮气净化的1 mol/L KOH溶液中进行. 0.6~1.0 V vs. RHE区域中出现明显的Pd氧化峰, 1.0~0.5 V vs. RHE则是PdO还原峰. 与PdNi/HfO
2和Pd/C相比, PdNi/HfO
2-O
v的PdO还原峰向低电位分别偏移了8 mV和14 mV (
图4b), 这是由于催化剂表面与OH
ads结合力增加, 有助于加速EG氧化
[25]. 此外, PdNi/HfO
2-O
v的PdO还原峰面积最大, 表明其参与催化的活性位点最多, 催化剂的电化学活性比表面积最大. 将PdO还原峰积分可得PdNi/HfO
2-O
v的电化学活性比表面积(ECSA)值为26.61 m
2/g分别是PdNi/HfO
2 (17.3 m
2/g)、Pd/C (8.6 m
2/g)的1.54、3.09倍(
图4c). 在1 mol/L KOH+1 mol/L EG中检测了乙二醇的氧化过程(
图4d), 在正向扫描的循环伏安(CV)曲线中出现吸附反应物的氧化峰, 而反向信号中的峰则对应于快速去除氧化过程生成的碳物质
[26]. PdNi/HfO
2-O
v催化剂具有最负的起始电位(0.3 V), 相比于PdNi/HfO
2和商业Pd/C催化剂分别减小了199 mV和167 mV, 表明其具有最低的EG氧化电位(
图4e). CV曲线负移可归因于氧空位的存在, 这增强了PdNi合金纳米颗粒与HfO
2之间的强相互作用, 进而形成了低电子密度的Pd. 这种缺电子结构有效降低了EGOR能垒, 导致氧化曲线的起始电位负移. 众所周知, Pd基合金催化剂易受CO毒化而使催化活性降低. 因此, 本工作在1 mol/L KOH电解质溶液中对所制备的材料进行抗CO毒化能力测试(支持信息图S4). PdNi/HfO
2-O
v的CO氧化峰电位位于0.78 V vs. RHE, 比PdNi/HfO
2 (0.81 V vs. RHE)和商业Pd/C (0.87 V vs. RHE)分别提前30 mV和90 mV, 显示出更优异的抗CO中毒能力, 这可归因于HfO
2载体中丰富的氧空位促进了催化剂在更低电位下进行CO
ads氧化. 此外, 本工作还总结了正向峰值电流密度(
If)与反向峰值电流密度(
Ib)之间的比值, 用以进一步反映催化剂的抗中毒能力. 如
图4f所示, PdNi/HfO
2-O
v (
If/
Ib=1.01)>PdNi/HfO
2 (
If/
Ib=0.92)>Pd/C (
If/
Ib=0.7). 综上所述, PdNi/HfO
2-O
v表现出优异的抗CO中毒能力, 这可归因于氧空位的存在及其独特的电子效应.
图4g显示, PdNi/HfO
2-O
v的峰值电流密度(MA)为10.28
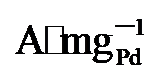
, 显著高于PdNi/HfO
2 (5.08
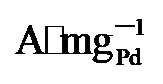
)和Pd/C (1.36
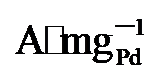
). 同时, PdNi/HfO
2-O
v的比活性(SA)达到38.6 mA•cm
-2, 相比于PdNi/HfO
2 (17.3 mA•cm
-2)和Pd/C (15.8 mA•cm
-2), 增强了2.23及2.55倍. 不仅如此, 本工作还将PdNi/HfO
2- O
v催化剂与前人报道的催化剂的EGOR性能进行了比较(
图4h)
[4,11,25,27-34], 结果显示, 本研究所制备的催化剂具有较好的电氧化性能.


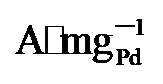 , 保留了86%的初始活性, 明显高于PdNi/HfO2 (3.32
, 保留了86%的初始活性, 明显高于PdNi/HfO2 (3.32 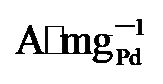 64.4%)和Pd/C (0.59
64.4%)和Pd/C (0.59 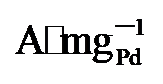 45%). 而在0.7 V vs. RHE下记录的2 h计时电流测试显示, PdNi/HfO2-Ov的电流密度趋于稳定且衰减最小. 质量活性保持最高(1.29
45%). 而在0.7 V vs. RHE下记录的2 h计时电流测试显示, PdNi/HfO2-Ov的电流密度趋于稳定且衰减最小. 质量活性保持最高(1.29 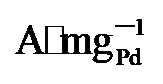 ), 分别是PdNi/HfO2 (0.68
), 分别是PdNi/HfO2 (0.68 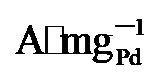 )和Pd/C (0.509
)和Pd/C (0.509 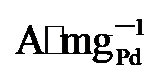 )的1.9、2.53倍. 由此可见, PdNi/HfO2-Ov具有较好的可重复性, 循环实用性的增加得益于氧空位的存在[35]. XPS结果显示Pd与HfO2载体间的电子转移可防止合金纳米颗粒与载体脱离, 维持结构稳定. 其次, 氧空位作为EG吸附及电子转移的活性位点, 能稳定EG分子从而增加电氧化反应的接触机会[36], 最终提升催化剂稳定性. 最后, 本工作在1 mol/L KOH+1 mol/L EG、-0.918 V vs. RHE的条件下进行了催化剂的电化学阻抗测试(EIS如图5d). Rs代表欧姆电阻, Rct代表电荷传递电阻, CPE则是双层电容[37]. 显然, PdNi/HfO2-Ov的阻抗弧直径小于PdNi/HfO2和Pd/C, 证明该催化剂在碱性乙二醇溶液中具有最小的电荷传质阻力和最高的电子转移速度[38]. 综上所述, PdNi/HfO2-Ov因其优越的导电性和较好的催化活性, 被视为一种极具潜力的DEGFCs阳极催化剂.
)的1.9、2.53倍. 由此可见, PdNi/HfO2-Ov具有较好的可重复性, 循环实用性的增加得益于氧空位的存在[35]. XPS结果显示Pd与HfO2载体间的电子转移可防止合金纳米颗粒与载体脱离, 维持结构稳定. 其次, 氧空位作为EG吸附及电子转移的活性位点, 能稳定EG分子从而增加电氧化反应的接触机会[36], 最终提升催化剂稳定性. 最后, 本工作在1 mol/L KOH+1 mol/L EG、-0.918 V vs. RHE的条件下进行了催化剂的电化学阻抗测试(EIS如图5d). Rs代表欧姆电阻, Rct代表电荷传递电阻, CPE则是双层电容[37]. 显然, PdNi/HfO2-Ov的阻抗弧直径小于PdNi/HfO2和Pd/C, 证明该催化剂在碱性乙二醇溶液中具有最小的电荷传质阻力和最高的电子转移速度[38]. 综上所述, PdNi/HfO2-Ov因其优越的导电性和较好的催化活性, 被视为一种极具潜力的DEGFCs阳极催化剂.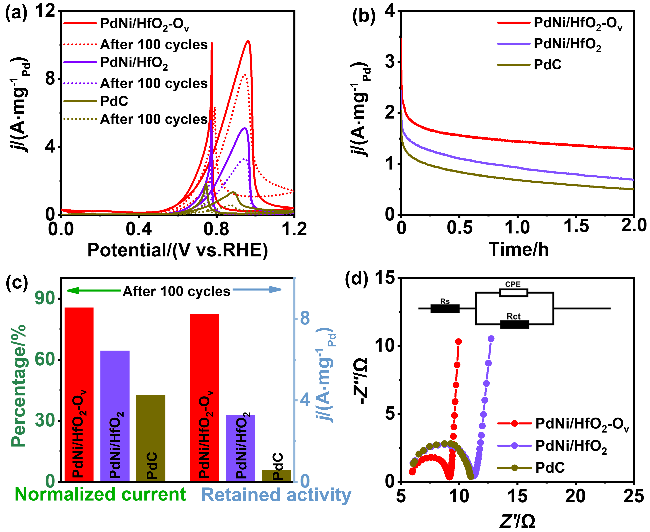
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
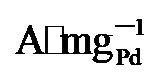 , 分别是PdNi/HfO2和商业Pd/C的2.02倍和7.56倍. 综上所述, 本工作为设计负载在缺陷型氧化物上的直接乙二醇燃料电池阳极催化剂提供了一种可行性策略.
, 分别是PdNi/HfO2和商业Pd/C的2.02倍和7.56倍. 综上所述, 本工作为设计负载在缺陷型氧化物上的直接乙二醇燃料电池阳极催化剂提供了一种可行性策略.