


1 引言
2 POCs的合成策略
2.1 不可逆键连接化学

图1 (a) D3h型有机笼Cage-2的化学结构[24]. (b)四棱柱型TPE基有机笼Cage-3的化学结构[25]. (c)四棱柱卟啉笼TPPCage8+的合成策略[27]Figure 1 (a) The chemical structure of D3h-organic cage Cage-2[24]. Copyright 2007 The American Chemical Society. (b) The chemical structure of quadrangular prismatic TPE-based cage Cage-3[25]. (c) The synthesis of tetragonal prismatic porphyrin cage TPPCage8+[27]. Copyright 2018 The American Chemical Society |
2.2 动态共价化学
2.2.1 硼酸酯缩合

图2 (a)立方八面体[12+8]型笼Cage-6a/b的合成[38]. (b)大型硼酸酯笼Cage-7的合成[39]. (c)直角棱柱型有机笼COP-5的合成[42]Figure 2 (a) The synthesis of cuboctahedral [12+8] cages Cage-6a/b[38]. Copyright 2014 Wiley-VCH. (b) The synthesis of giant boronic ester cage Cage-7[39]. Copyright 2021 Wiley-VCH. (c) The synthesis of rectangular prismatic cage COP-5[42]. Copyright 2011 The American Chemical Society |
2.2.2 烯烃/炔烃复分解
2.2.3 亚胺缩合

图3 (a)八面体笼Cd6La12的合成[48]. (b) [12+8]型有机笼Cage-9的合成[49]. (c) [4+4]型有机笼Cage-12的合成[52]. (d)由X射线晶体学确定的CC1、CC2及CC3的结构图[45]. (e)有机笼Cage-11a/b的合成[51]Figure 3 (a) The synthesis of octahedral cage Cd6La12[48]. Copyright 2011 The Royal Society of Chemistry. (b) The synthesis of [12+8] organic cage Cage-9[49]. Copyright 2013 The Royal Society of Chemistry. (c) The synthesis of [4+4] organic cage Cage-12[52]. Copyright 2018 Wiley-VCH. (d) Structure diagram of CC1, CC2 and CC3 determined by X-ray crystallography[45]. (e) The synthesis of organic cages Cage-11a/b[51] |
2.3 合成策略与性能关联
2.3.1 孔隙率
2.3.2 稳定性
2.3.3 合成产率
3 催化能源转换
3.1 析氢反应(HER)
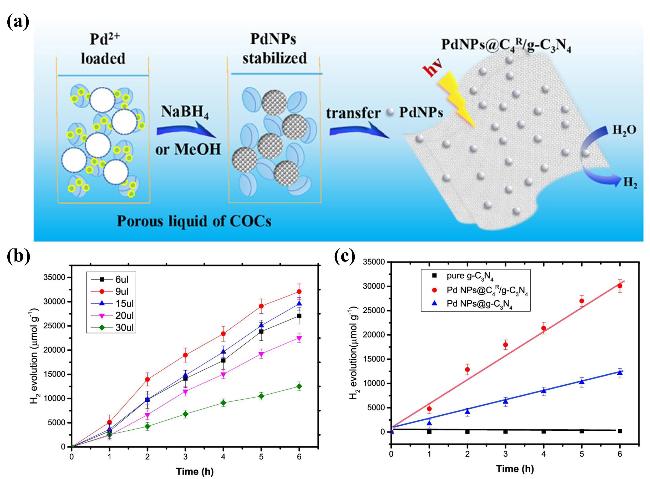
图4 (a)用于光催化析氢的PdNPs@C4R/g-C3N4复合材料的构建示意图. (b)不同Pd纳米颗粒负载量时g-C3N4光催化活性对比. (c)不同催化剂的光催化产氢速率对比[56]Figure 4 (a) Schematic illustration of the construction of PdNPs@C4R/g-C3N4 composite for photocatalytic hydrogen evolution. (b) The comparison of H2 production for g-C3N4 with different Pd NP loading amounts. (c) The comparison of photocatalytic hydrogen evolution rate for different catalysts[56]. Copyright 2020 The American Chemical Society |
3.2 氧还原反应(ORR)
3.3 硝酸根还原反应(NO3RR)
3.4 二氧化碳还原反应(CO2RR)

图5 (a)光致变色笼TAE-DTE的光异构体[64]. (b) TAE-DTE-O在可见光(左)和全范围(右)光照下的光催化CO2还原表现[64]. (c) FePB-2(P)催化剂的设计策略[66]. (d) FePB-2(P)在CO2饱和CH3CN溶液中的光催化活性[66]. (e)不同催化剂在CO2饱和的DMF溶液中的催化TON值对比(以Fe位点为准)[66]Figure 5 (a) Photo-isomers of the photochromic organic cage TAE-DTE[64]. (b) The photocatalytic CO2 reduction performance of TAE-DTE-O under visible light (left) and full range light (right)[64]. Copyright 2021 The Royal Society of Chemistry. (c) The design strategy of catalyst FePB-2(P)[66]. (d) The catalytic activity of FePB-2(P) in CO2-saturated CH3CN[66]. (e) TON per[Fe] comparison of photocatalytic CO2RR for different catalysts in CO2-saturated DMF[66]. Copyright 2022 Wiley-VCH |

图6 (a)流动电池的工作示意图. (b) Cu-nr/CC3和Cu-nr在不同电位下的电流密度对比. (c) Cu-nr/CC3和Cu-nr在不同电位下C2+产物的法拉第效率对比. (d) Cu-nr/CC3和Cu-nr在不同电位下H2产物的法拉第效率对比. (e)不同催化剂在-0.9 V电位下的产物法拉第效率对比[69]Figure 6 (a) Schematic diagram of the flow-cell. (b) The comparison of current density between Cu-nr/CC3 and Cu-nr at different potentials. (c) The FE of C2+ products and (d) H2 at different potentials over Cu-nr/CC3 and Cu-nr. (e) Comparison of FE of products over different catalysts at -0.9 V potential[69]. Copyright 2022 Wiley-VCH |
3.5 有机转换反应

图7 (a) 通过向卟啉有机笼中引入Au团簇调节活性氧物种的示意图. (b) Au⊂TPPCage•Cl和TPPCage•Cl催化CEES氧化的产率对比. (c) Au⊂TPPCage•POM双催化功能示意图. (d) Au⊂TPPCage•POM和K8[Nb6O19]用于DECP水解的产率对比[74].Figure 7 (a) The schematic diagram of Au clusters introducing porphyrin cages to regulate ROS. (b) The yield of CEES oxidation comparison of Au⊂TPPCage•Cl and TPPCage•Cl. (c) Schematic illustration of the dual catalytic functionality of Au⊂TPPCage•POM. (d) The yield of DECP hydrolysis comparison of Au⊂TPPCage•POM and K8[Nb6O19][74]. Copyright 2024 Science China Press |
4 能量存储
4.1 锂电池

图8 (a)由RCC1-Cl到多孔有机笼电解质Li-RCC1-ClO4的合成步骤[14]. (b)不同LiClO4含量的Li-RCC1-ClO4 SSE离子电导率对比[14]. (c) PEO-LiClO4和PEO/Li-RCC1-ClO4的LSV曲线对比[14]. (d) C60@POC的主客体结构示意图[82]. (e)在光照/非光照条件下C60@POC的充放电曲线[82]. (f)在光照/非光照下POC和C60@POC的Nyquist图对比[82]Figure 8 (a) The synthetic procedure of the porous cage electrolyte Li-RCC1-ClO4 from RCC1-Cl[14]. (b) Ion conductivity of the Li-RCC1-ClO4 SSE with different LiClO4 contents[14]. (c) The LSV results comparison of PEO-LiClO4 and PEO/Li-RCC1-ClO4[14]. Copyright 2022 Springer Nature. (d) The guest-host structure of C60@POC[82]. (e) Charge/discharge curves of C60@POC with/without irradiation[82]. (f) The Nyquist plots comparison of POC and C60@POC with/without irradiation[82]. Copyright 2022 The Royal Society of Chemistry |

图9 (a) ⅡC-1@Na2S5的封装步骤示意图[87]. (b) ⅡC-1@Na2S5 LSB的倍率性能[87]. (c) ⅡC-1@Na2S5 LSB的循环性能及库伦效率[87]. (d)装载CC3/PP和CC3隔膜的电池在1 C下循环500次的Li+和多硫化物离子筛分选择性和循环性能对比[86]. (e) CC3/PP和PP隔膜在不同库伦速率下的倍率性能[86]Figure 9 (a) The encapsulation procedure of ⅡC-1@Na2S5[87]. (b) The rate performance and (c) cycling performance and coulombic efficiency for ⅡC-1@Na2S5 LSB[87]. Copyright 2022 Elsevier. (d) The comparison of the Li+ and polysulfide ion sieving selectivity and cycling performance of batteries equipped with CC3/PP and bare PP separators at 1 C over 500 cycles[86]. (e) Rate performance at various C-rates for CC3/PP and PP[86]. Copyright 2022 The American Chemical Society |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}