

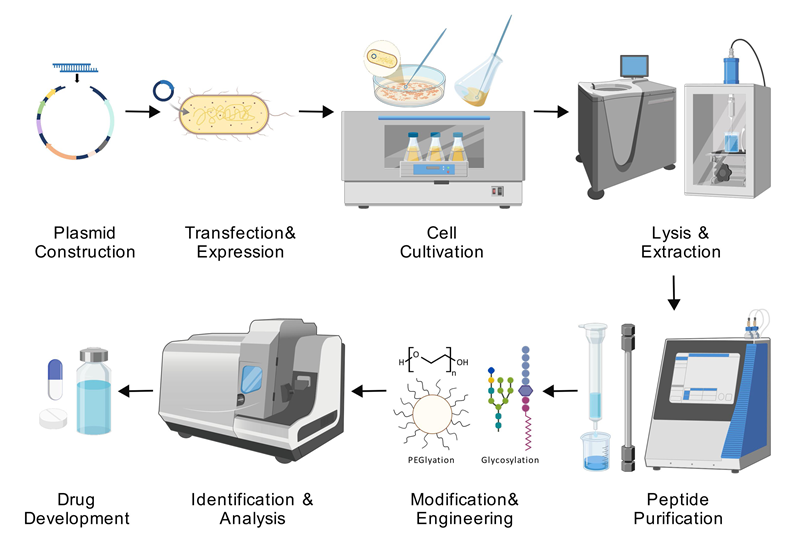
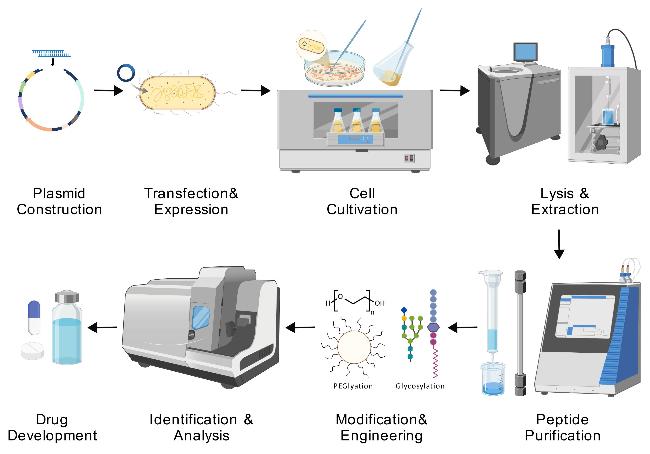
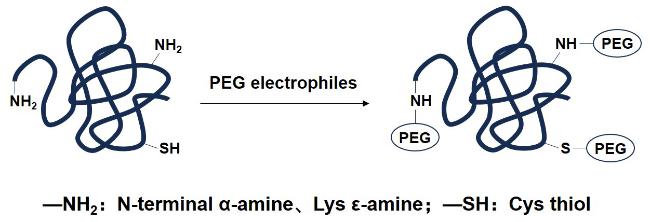
1 引言
表1 已上市或处于临床试验的毒素肽药物Table 1 Toxin-derived peptide drugs approved or under clinical trials |
| 名称 | 来源 | 用药方式 | 作用靶点/机制 | 主要适应症 | 临床状态 | 备注 |
|---|---|---|---|---|---|---|
| Cobratide (克痛宁) | 中华眼镜蛇 (Naja atra) | 静脉注射/口服 | 烟碱型乙酰胆碱受体阻断剂 | 慢性关节痛、坐骨神经痛、神经性 头痛 | 已上市 | 国药准字H53022101[27] |
| Ziconotide (Prialt®) | Conus magus | 鞘内注射 | Cav2.2通道 阻断剂 | 严重慢性疼痛 | 已上市 | New Drug Application (NDA): 021060[28] |
| μ-TRTX-Hhn1b (Halneuron) | Ornithoctonus hainana | 皮下注射 | 电压门控钠通道 Nav1.7抑制剂 | 化疗性神经性疼痛 | II期临床 | ClinicalTrials.gov ID: NCT06848348[29] |
| Tetrodotoxin | pufferfish | 肌内注射/皮下注射 | 电压门控钠通道 Nav阻断剂 | 中度或重度癌症疼痛、化疗性神经性疼痛 | Ⅱ/Ⅲ期临床 | ClinicalTrials.gov ID: NCT00726011/NCT00725114/NCT01655823[29] |
| ACV-1 (a-Vc1.1) | Conus victoriae | 皮下注射 | GABAB受体 激活剂 | 糖尿病性神经病变疼痛 | II期临床 | 因缺乏疗效而停止[30] |
| Contulakin-G (CGX-1160) | Conus geographus | 鞘内注射 | 神经降压素受体激活剂 | 神经病理性疼痛 | II期临床 | 因开发商公司破产而 停止[30] |
| Leconotide (ω-conotoxin CVID) | Conus catus | 鞘内注射 | Cav2.2通道 阻断剂 | 神经病理性疼痛 | Ⅰ/II期临床 | 因开发商公司破产而 停止[30] |
| Xen 2174 (c-CTX MrIA) | Conus marmoreus | 鞘内注射 | 作用于去甲肾上腺素转运体 | 术后疼痛 | II b期临床 | 已停止[30] |
2 ASICs的结构与功能
2.1 ASIC的分子结构
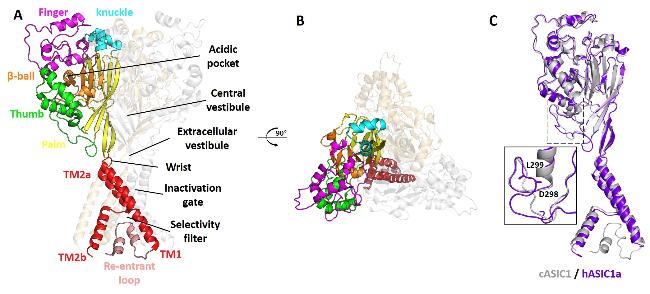
图1 ASIC通道的分子结构 (A)静息状态cASIC1三聚体侧面构象(PDB号: 6vtl); (B)静息状态cASIC1三聚体俯视构象; (C) cASIC1(灰色, PDB号: 6vtl)与hASIC1a(紫色, PDB号: 7cfs)单亚基结构差异对比Figure 1 Molecular structure of the ASIC channel (A) Lateral conformation of the resting state cASIC1 trimer (PDB ID: 6vtl); (B) Top view conformation of the resting state cASIC1 trimer; (C) Comparison of the structural differences between the single subunit structures of cASIC1 (grey, PDB ID: 6vtl) and hASIC1a (purple, PDB ID: 7cfs) |
2.2 ASIC的门控机制
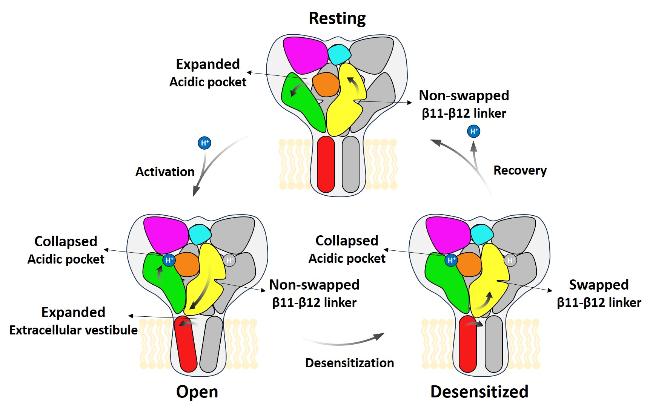
2.3 ASICs的病理功能与靶向优势
3 毒素肽的化学特性与靶向机制
3.1 代表性毒素肽的化学结构
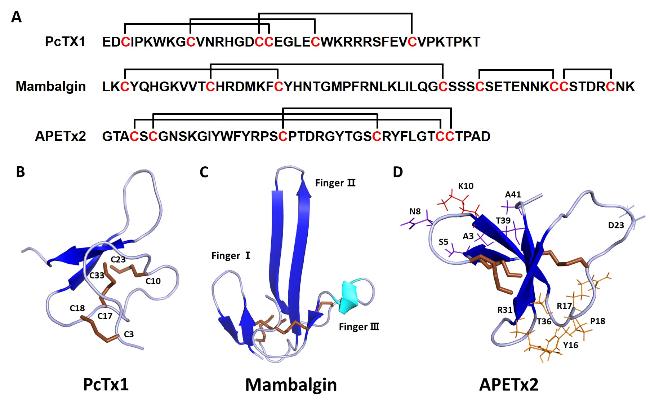
图3 (A)三种代表性毒素肽的氨基酸序列, 各肽二硫键已标出; (B~D)三种代表性毒素肽的结构, 其中青色为α螺旋, 蓝色为β折叠, 褐色为二硫键(PBD号: PcTx1-3s3x, Mamb-5dz5, APETx2-2mub)Figure 3 (A) Sequences of three representative toxin peptides, with the disulphide bond of each peptide indicated; (B~D) Structures of three representative toxin peptides, with α helices in cyan, β folds in blue and disulphide bonds in brown (PBD ID: PcTx1-3s3x, Mamb-5dz5, APETx2-2mub) |
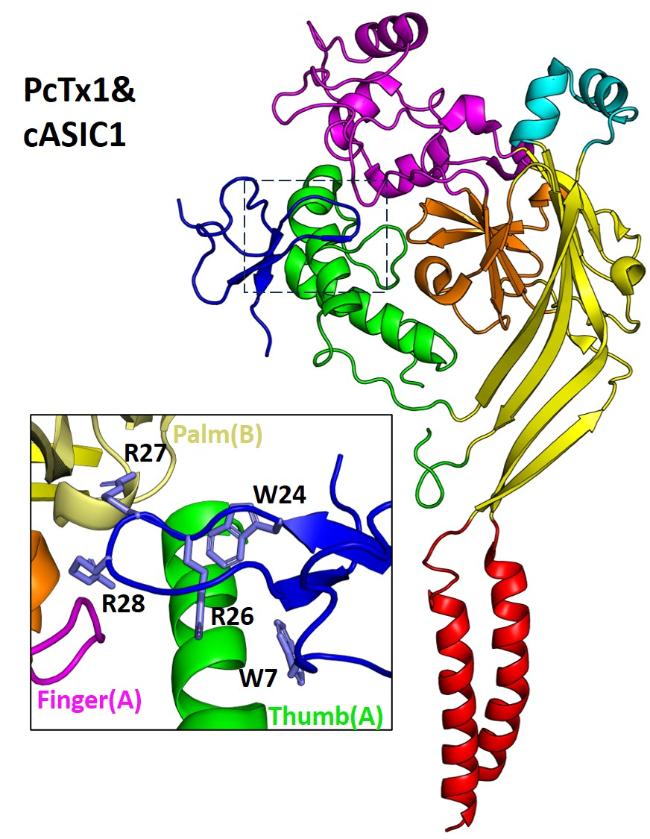
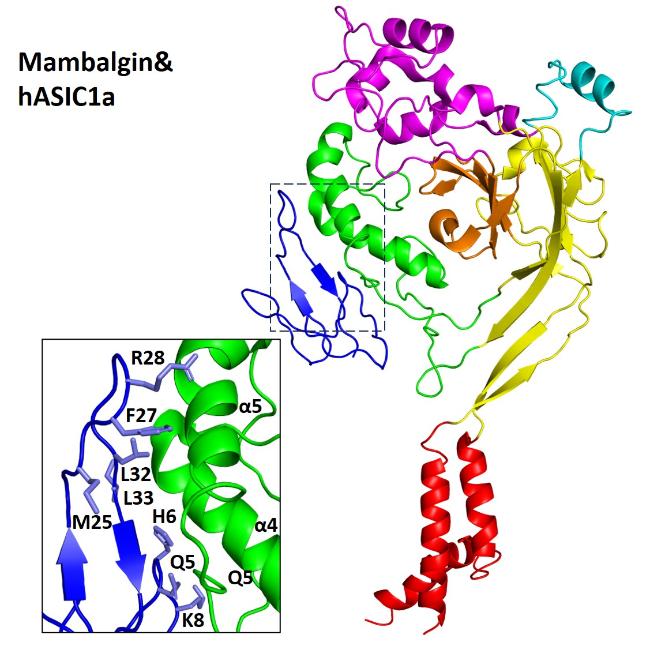
3.2 代表性毒素肽与ASICs的相互作用机制
表2 靶向ASIC通道的代表性毒素肽Table 2 Representative toxin peptides targeting ASIC channels |
| 毒素名称 | 来源生物 | 靶向ASIC亚型 | 调节方式 | 作用特点 |
|---|---|---|---|---|
| APETx2 | 海葵 | ASIC3 | 抑制 | 可能与孔阻塞相关, 非pH依赖 |
| Hi1a | 蜘蛛 | ASIC1a | 抑制 | PcTx1的串联体, 更强效长效 |
| Mambalgin | 曼巴蛇 | ASIC1a、1b | 抑制 | 干扰亚基构象转变, pH依赖 |
| MitTx | 珊瑚蛇 | ASIC1a | 激活 | 稳定通道开放构象, 组成型激活 |
| PcTx1 | 蜘蛛 | ASIC1a | 抑制 | 稳定通道脱敏构象, pH依赖 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}