


1 引言
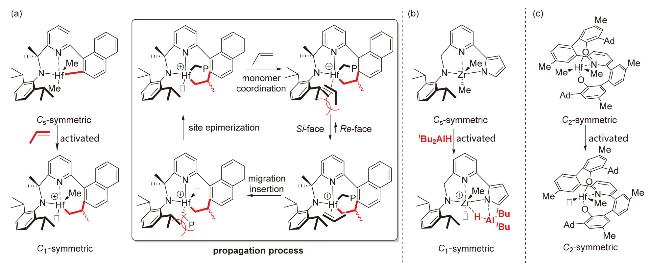
2 结果与讨论
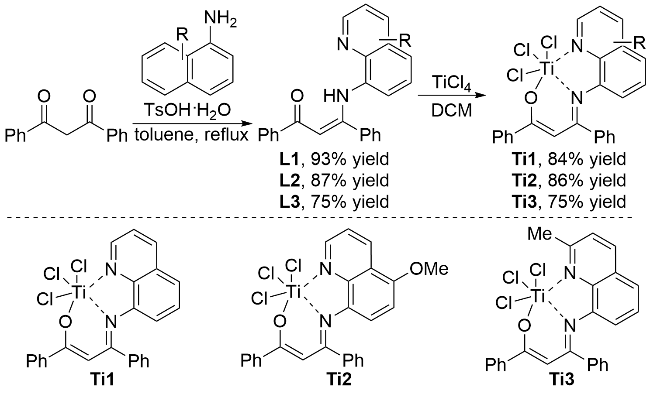
2.1 催化剂选择与合成
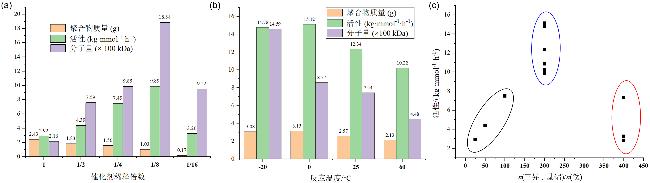
2.2 聚合反应方法学研究
2.2.1 1-丁烯均聚反应
表1 钛催化1-丁烯聚合Table 1 1-Butene polymerization with titanium catalysts |
| Entry | Conditionsa | 1-Butene/[Al]/[Ti]/[B] (mmol) | Yield (g) /conv. (%) | Activity/ (kg•mmol−1•h−1) | Mnb/ kDa | PDIb | Isotacticityc (% mmmm) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ti1/AlEt3/[Ph3C][B(C6F5)4]/tol/25 ℃ | 86/0.25/0.01/0.01 | 2.24/47 | 2.69 | 27 | 3.1 | 82 | ||||||||
| 2 | Ti1/AlEt3/B(C6F5)3/tol/25 ℃ | 86/0.25/0.01/0.01 | 0.41/9 | 0.49 | 33 | 2.8 | 81 | ||||||||
| 3d | Ti1/AlEt3/[Ph3C][B(C6F5)4]/DCM/25 ℃ | 86/0.25/0.01/0.01 | 1.30/27 | 1.56 | 27 | 2.8 | 80 | ||||||||
| 4 | Ti1/AlMe3/[Ph3C][B(C6F5)4]/tol/25 ℃ | 86/0.25/0.01/0.01 | n.p./0 | n.r. | — | — | — | ||||||||
| 5 | Ti1/AliBu3/[Ph3C][B(C6F5)4]/tol/25 ℃ | 86/0.25/0.01/0.01 | 2.43/51 | 2.92 | 216 | 3.3 | 83 | ||||||||
| 6 | Ti1/AliBu3/[Ph3C][B(C6F5)4]/tol/25 ℃ | 86/0.25/0.005/0.01 | 1.83/38 | 4.39 | 759 | 2.4 | 83 | ||||||||
| 7 | Ti1/AliBu3/[Ph3C][B(C6F5)4]/tol/25 ℃ | 86/0.25/0.0025/0.01 | 1.56/33 | 7.49 | 985 | 3.1 | 83 | ||||||||
| 8 | Ti1/AliBu3/[Ph3C][B(C6F5)4]/tol/25 ℃ | 86/0.25/0.00125/0.01 | 1.03/21 | 9.89 | 1884 | 1.9 | 80 | ||||||||
| 9 | Ti1/AliBu3/[Ph3C][B(C6F5)4]/tol/25 ℃ | 86/0.25/0.000625/0.01 | 0.17/4 | 3.26 | 952 | 2.9 | 84 | ||||||||
| 10 | Ti1/AliBu3/[Ph3C][B(C6F5)4]/tol/25 ℃ | 86/0.5/0.0025/0.01 | 2.57/54 | 12.34 | 744 | 2.6 | 83 | ||||||||
| 11 | Ti1/AliBu3/[Ph3C][B(C6F5)4]/tol/25 ℃ | 86/0.5/0.00125/0.01 | 0.76/16 | 7.30 | 1489 | 1.8 | 83 | ||||||||
| 12 | Ti1/AliBu3/[Ph3C][B(C6F5)4]/tol/25 ℃ | 86/1/0.0025/0.01 | 0.59/12 | 2.83 | 1062 | 2.2 | 84 | ||||||||
| 13 | Ti1/AliBu3/[Ph3C][B(C6F5)4]/tol/60 ℃ | 86/0.5/0.0025/0.01 | 2.13/44 | 10.22 | 448 | 3.9 | 82 | ||||||||
| 14 | Ti1/AliBu3/[Ph3C][B(C6F5)4]/tol/0 ℃ | 86/0.5/0.0025/0.01 | 3.15/66 | 15.12 | 857 | 1.8 | 84 | ||||||||
| 15 | Ti1/AliBu3/[Ph3C][B(C6F5)4]/tol/-20 ℃ | 86/0.5/0.0025/0.01 | 3.08/64 | 14.78 | 1459 | 1.9 | 81 | ||||||||
| 16 | Ti2/AliBu3/[Ph3C][B(C6F5)4]/tol/0 ℃ | 86/0.5/0.0025/0.01 | 2.26/47 | 10.85 | 1017 | 2.1 | 81 | ||||||||
| 17e | Ti3/AliBu3/[Ph3C][B(C6F5)4]/tol/0 ℃ | 86/0.5/0.0025/0.01 | n.p./0 | n.r. | — | — | — | ||||||||
| 18e | Ti3/AliBu3/[Ph3C][B(C6F5)4]/tol/25 ℃ | 86/0.5/0.0025/0.01 | n.p./0 | n.r. | — | — | — | ||||||||
a Polymerization conditions: 1-Butene (4.8 g, 86 mmol), Ti catalyst (in 2 mL toluene), AlR3, co-catalyst (0.01 mmol in 2 mL toluene), -20~60 ℃, 5 min, in a sealed tube, quenched by acidified ethanol until the set time. b Determined by GPC. c Determined by quantitative 13C NMR spectrum. d Replace the solvent of catalyst and co-catalyst with dichloromethane instead of toluene. e The reaction time was 60 min. |
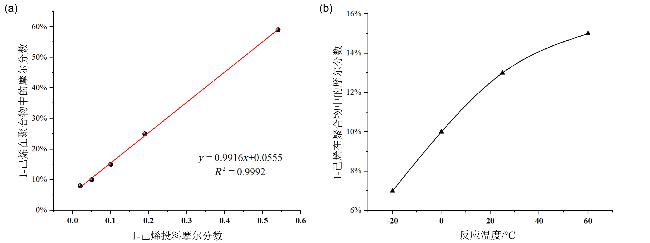
2.2.2 1-丁烯与1-己烯共聚反应
表2 钛催化1-丁烯与1-己烯共聚aTable 2 1-Butene/1-hexene copolymerization with titanium catalystsa |
| Entry | Co-Monomer (mmol) | T/℃ | Yield/g | Activity/ (kg•mmol−1•h−1) | Mnb/ kDa | PDIb | x/w c (%) |
|---|---|---|---|---|---|---|---|
| 1 | 1-hexene (2) | 0 | 2.06 | 9.89 | 1340 | 2.2 | 8/11 |
| 2 | 1-hexene (5) | 0 | 2.41 | 11.57 | 1441 | 2.0 | 10/14 |
| 3 | 1-hexene (10) | 0 | 2.20 | 10.56 | 1599 | 2.2 | 15/21 |
| 4 | 1-hexene (20) | 0 | 2.13 | 10.22 | 1491 | 2.3 | 25/33 |
| 5 | 1-hexene (100) | 0 | 3.95 | 18.96 | 2637 | 2.1 | 59/68 |
| 6 | 1-hexene (5) | -20 | 3.24 | 15.55 | 1454 | 2.1 | 7/10 |
| 7 | 1-hexene (5) | 25 | 2.38 | 11.42 | 952 | 2.7 | 13/18 |
| 8 | 1-hexene (5) | 60 | 1.99 | 9.55 | 849 | 3.5 | 15/21 |
a Polymerization conditions: 1-Butene (4.8 g, 86 mmol), 1-hexene, Ti1 (0.0025 mmol in 2 mL toluene), AliBu3 (0.5 mL, 1 mol•L−1 in toluene), [Ph3C][B(C6F5)4] (0.01 mmol in 2 mL toluene), -20~60 ℃, 5 min, in a sealed tube, quenched by acidified ethanol until the set time. b Determined by GPC. c Determined by 1H NMR spectrum. |
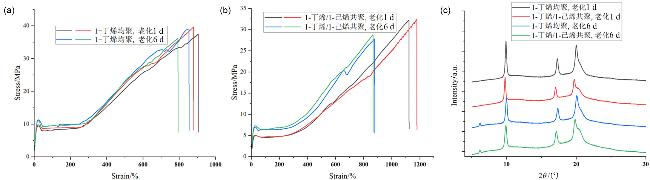
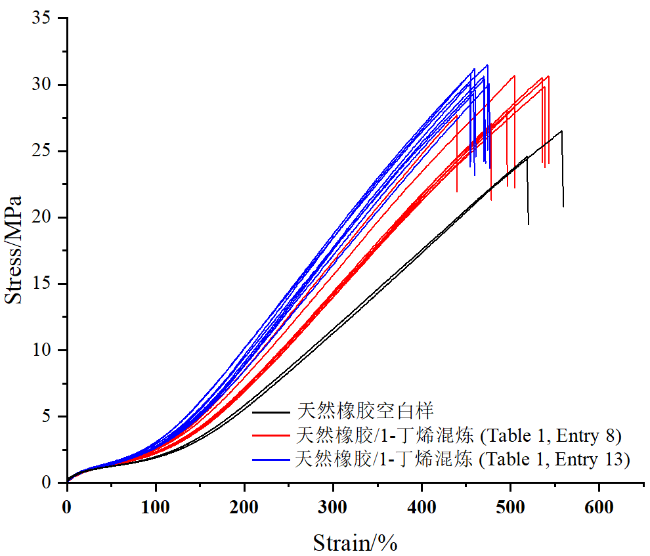
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}