

1 引言
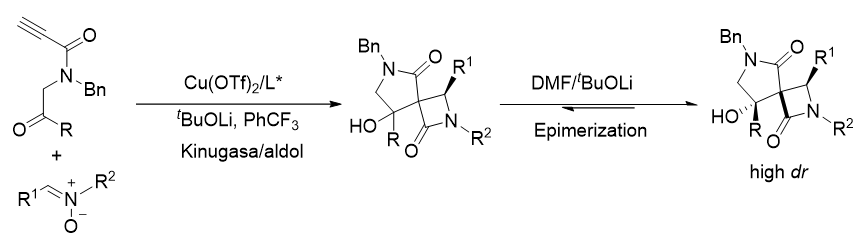
近年来, 具有环张力的手性骨架在药物研发领域受到了广泛的关注. 其中β-内酰胺以及螺环β-内酰胺类化合物因其独特的结构和显著的生物活性, 引起了合成化学家和药物化学家极大的研究兴趣. 这类结构作为许多生物活性分子和天然产物中的关键结构单元, 展现出广泛的生物活性, 包括抗肿瘤、抗HIV、抗菌和抗疟疾活性等(图1)[1]. 鉴于其重要性, 研究者们投入了大量精力开发合成这类结构的新方法和新策略[2]. 例如, 基于经典的[2+2]亚胺和烯酮的环加成反应(Staudinger β-内酰胺合成)[3]以及N-杂环卡宾(NHC)催化的环加成反应等, 已被广泛用于高效合成β-内酰胺结构, 并通过进一步后续转化或串联反应等构建螺环β-内酰胺化合物[4]. 然而, 尽管β-内酰胺以及螺环β-内酰胺化合物的合成已取得显著进展, 开发更加温和、高效的方法来直接构建螺环β-内酰胺结构, 仍然是当前研究的重要方向.
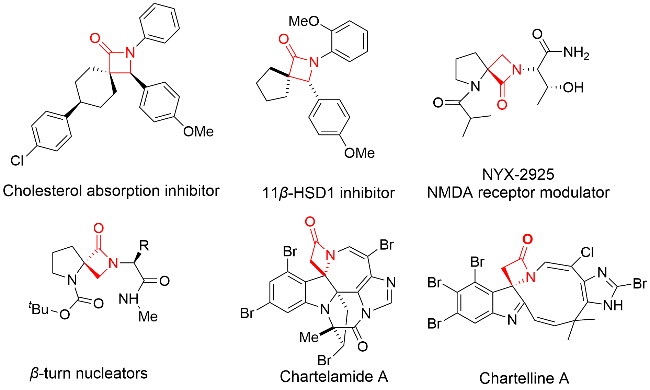
在众多合成β-内酰胺的策略中, 铜催化的Kinugasa反应[5]因其简单高效而备受关注. 该反应利用一价铜盐催化端炔与硝酮的环加成反应, 直接生成β-内酰胺. 这一反应最早由Kinugasa和Hashimoto[6]于1972年报道, 并因此得名为Kinugasa反应. 1995年, Miura等[7]首次实现了不对称Kinugasa反应. 此后该反应被广泛应用于手性β-内酰胺的合成[8-9]. 随着研究的深入, Kinugasa反应的机理也逐渐明晰: 一般认为, 反应首先形成端炔铜盐, 随后与硝酮通过[3+2]环加成生成不稳定的五元环中间体, 进一步重排为四元环的烯醇亚铜中间体, 最终通过质子化生成β-内酰胺产物[10].
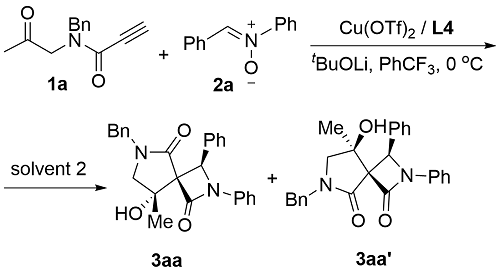
我们课题组[15]采用丙炔酰胺类化合物作为端炔底物, 成功发展了一系列新型中断Kinugasa反应. 这类反应中生成的烯醇铜中间体α位带有一个吸电子的酰基, 显著增强了中间体的稳定性. 此外, 在碱性条件下, 中间体可通过质子化-去质子化的平衡过程, 与亲电试剂发生反应. 因此, 在丙炔酰胺类化合物与硝酮的Kinugasa反应中, 我们能够引入多种试剂或官能团, 实现了多样化的串联反应, 极大地拓展了这类反应的应用范围. 例如我们使用邻碘代芳基作为亲电基团, 实现了Kinugasa/C—C偶联串联反应; 以及采用α,β-不饱和酯作为亲电基团, 成功发展了Kinugasa/Michael加成串联反应; 2023年, 我们课题组[15b]进一步采用羰基作为亲电基团, 在分子内捕捉Kinugasa反应中间体, 首次成功实现了不对称Kinugasa/Aldol串联反应(图2b). 这类反应中使用邻位羰基芳胺连接的丙炔酰胺作为底物与硝酮反应, 能够以高对映选择性和立体选择性构建具有三个连续立体中心的手性螺环β-内酰胺-二氢喹啉酮结构. 在这类反应中, 由于产物中苯环以及两个酰胺结构的存在, 在碱性条件下非常容易通过retro-aldol/aldol过程, 生成构型单一的稳定产物. 因此, 反应的非对映选择性非常高, 大多情况下只能观察到单一非对映异构体.
在我们的工作发表后不久, Lautens等[14]也报道了类似的铜催化不对称Kinugasa/Aldol串联反应(图2c). 在他们的体系中, 采用酮连接的端炔与硝酮反应, 以三乙胺为碱, 在乙腈中生成手性螺环β-内酰胺-吡咯烷酮产物, 同样表现出良好的对映选择性, 但其非对映选择性不尽如人意. 在本工作中, 我们基于前期不对称Kinugasa/Aldol反应研究基础, 进一步拓展了反应底物范围, 采用烷基酮连接的丙炔酰胺类底物与硝酮反应, 成功合成了手性螺环β-内酰胺产物. 然而, 与Lautens的研究结果类似, 反应的非对映选择性并不理想. 我们分析认为, 尽管该反应与我们2023年报道的Kinugasa/Aldol串联反应在形式上相似, 但反应产物的性质存在显著差异. 在我们2023年的工作中, 螺环β-内酰胺-二氢喹啉酮结构由于羟基邻位芳环的存在, 使得羟基具有较高的反应活性, 易于通过retro-aldol/aldol过程生成单一构型的稳定产物, 从而表现出优异的非对映选择性. 而在本文研究中, 采用烷基酮连接的丙炔酰胺类底物时, 产物中羟基活泼性显著降低, 难以通过retro-aldol/aldol过程实现单一构型产物的高效生成. 为了提高其非对映选择性, 我们提出了一种可能的解决方案: 在反应完成后, 改用极性更大的溶剂, 如N,N-二甲基甲酰胺(DMF)以促进retro-aldol/aldol异构化的过程, 从而提升产物的非对映选择性(图2d). 在此, 我们将对这一研究工作进行详细介绍.
2 结果与讨论
2.1 反应条件筛选
我们以N-苄基-N-(2-氧代丙基)丙炔酰胺1a与N-1-二苯基硝基酮2a的反应为模型案例展开研究. 如表1所示, 反应首先以Cu(OTf)2为催化剂、tBuOLi为碱, 在PhCF3溶剂中筛选了多种手性双噁唑啉配体(L1~L5). 在这些反应中, 主要分离得到两种非对映异构体3aa及3aa'. 研究发现, 带有边臂的双噁唑啉配体(L2, L3, L5)相较于不带边臂的配体(L1, L4), 能够显著提高产物的非对映选择性, 但主要产物3aa的对映选择性却有所降低(表1, Entries 1~5). 其中, 使用配体L2时, 非对映体比例最高, 达到10∶1, 但主要非对映体3aa的对映选择性差(er=53∶47, 表1, Entry 2). 而使用配体L4时, 尽管非对映选择性仅为3∶1(表1, Entry 4), 但主要产物3aa的对映选择性最高(er=90∶10, 表1, Entry 4), 且反应收率也达到66%. 因此, 我们选择L4作为配体, 进一步对溶剂和碱进行优化. 在溶剂筛选过程中, 我们发现在1,2-二氯乙烷(DCE)、四氢呋喃(THF)、二氧六环、CH3CN等溶剂中, 反应的主要产物收率及对映选择性均有所下降(表1, Entries 6~9), 不如在PhCF3溶剂中的效果理想. 值得注意的是, 在DMF中, 完全没有目标产物生成, 这可能是由于硝酮原料在DMF中不稳定, 容易分解所致(表1, Entry 10). 此外, 我们还尝试了不同强度的碱(如K2CO3、Cs2CO3和NaH), 发现它们均能促进反应进行, 但效果都不及tBuOLi.
表1 反应条件优化aTable 1 Optimization of reaction conditionsa |
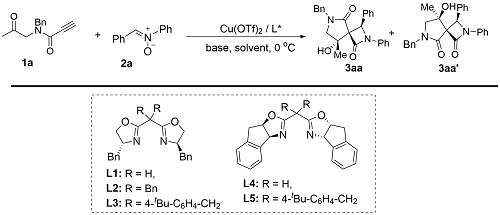
| Entry | L* | Solvent | Base | Yieldb/% | drc (3aa/3aa') | erd (3aa) |
|---|---|---|---|---|---|---|
| 1 | L1 | PhCF3 | tBuOLi | 41 | 4∶1 | 62∶38 |
| 2 | L2 | PhCF3 | tBuOLi | 39 | 10∶1 | 53∶47 |
| 3 | L3 | PhCF3 | tBuOLi | 58 | 8∶1 | 55∶45 |
| 4 | L4 | PhCF3 | tBuOLi | 66 | 3∶1 | 90∶10 |
| 5 | L5 | PhCF3 | tBuOLi | 58 | 7∶1 | 55∶45 |
| 6 | L4 | MeCN | tBuOLi | 51 | 1∶1 | 53∶47 |
| 7 | L4 | DCE | tBuOLi | 36 | 6∶1 | 64∶36 |
| 8 | L4 | 1,4-dioxane | tBuOLi | 47 | 2∶1 | 86∶14 |
| 9 | L4 | THF | tBuOLi | 41 | 3∶1 | 88∶12 |
| 10 | L4 | DMF | tBuOLi | n.d. | — | — |
| 11 | L4 | PhCF3 | K2CO3 | 46 | 3∶1 | 80∶20 |
| 12 | L4 | PhCF3 | Cs2CO3 | 44 | 2∶1 | 85∶15 |
| 13 | L4 | PhCF3 | NaH | 35 | 3∶1 | 75∶25 |
a Reaction conditions: 1a (0.2 mmol), 2a (0.24 mmol), Cu(OTf)2 (10 mol%), ligand (12 mol%), base (0.3 mmol), solvent (2 mL), 0 ℃, 12 h. b Isolated yields of 3aa/3aa'. c Determined by 1H NMR analysis of the crude reaction mixture. d Determined by HPLC analysis. |
考虑到在我们2023年所发展的Kinugasa/Aldol串联反应体系中, 生成的螺环β-内酰胺-二氢喹啉酮结构中, 由于aldol缩合形成的羟基邻位存在芳环, 活性较高, 易于通过retro-aldol/aldol过程实现高非对映选择性产物的生成. 而在本研究中, 螺环产物3aa/3aa', 羟基邻位为甲基, 因此在PhCF3作为溶剂时, 羟基的活性较低, 难以通过retro-aldol/aldol过程提高非对映选择性. 为了促进非对映体富集过程, 我们把研究重点放在第二步反应条件的优化. 如表2所示, 通过筛选不同的溶剂, 以期实现更高的非对映选择性. 具体操作如下: 在tBuOLi促进反应完成后, 除去溶剂PhCF3, 向体系中加入待筛选的溶剂, 并在室温下搅拌5 h. 实验结果表明, DMF能够有效促进异构化反应, 主要产物的产率可接受, 且非对映选择性得到显著提高(表2, Entry 6).
表2 反应条件进一步优化aTable 2 Further optimization of reaction conditionsa |
| Entry | Solvent 2 | Yieldb/% | drc (3aa/3aa') |
|---|---|---|---|
| 1 | PhCF3 | 46 | 3∶1 |
| 2 | DCE | 36 | 4∶1 |
| 3 | THF | 38 | 3∶1 |
| 4 | MeCN | 41 | 6∶1 |
| 5 | 1,4-dioxane | 46 | 3∶1 |
| 6 | DMF | 51 | 9∶1 |
a Reaction conditions: 1a (0.2 mmol), 2a (0.24 mmol), Cu(OTf)2 (10 mol%), L4 (12 mol%), tBuOLi (0.3 mmol), PhCF3 (2 mL), 0 ℃, 12 h. then evaporated the PhCF3 and added solvent 2, for 5 h at room temperature. b Isolated yields of 3aa/3aa'. c Determined by 1H NMR analysis of the crude reaction mixture. |
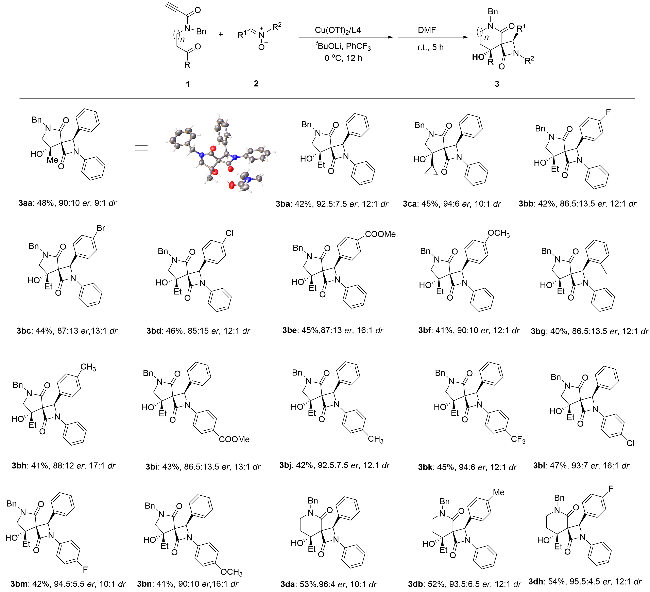
2.2 底物范围
对反应进行条件优化后, 我们进一步探索了反应的底物适用范围. 结果如图3所示. 首先, 我们考察了不同的烷基酮连接的丙炔酰胺底物. 研究发现, 末端为甲基、乙基或环丙基的酮连接的丙炔酰胺底物1a~1c均能顺利参与反应, 以中等收率和良好的非对映选择性生成相应的产物3aa~3ca, 且主要产物的对映选择性较高. 接下来, 我们进一步研究了硝酮的适用范围. 结果表明, 硝酮的两个芳基环上不同的取代基, 如卤素、甲基、甲氧基、三氟甲基、酯基等, 在我们的反应条件下, 均具有良好的耐受性, 能以中等的收率生成相应的产物3bb~3bn, 并表现出良好的非对映选择性和对映选择性. 此外, 在相同条件下, 延长炔酰胺底物中的碳链, 生成相应的4,6-螺环产物3da, 3db和3dh. 这些产物的非对映选择性良好, 而对映选择性略高于相应的4,5-螺环. 为了确定产物的绝对构型, 我们对产物3aa进行了X-单晶衍射实验, 确定了其绝对构型. 并以此为基础通过类比推断了其他产物的绝对构型. 此外, 我们还分离了非主要异构体3aa', 并通过X-单晶衍射实验确定了其绝对构型[16].
2.3 控制实验
为了更好地理解反应过程, 我们开展了一系列对照实验, 结果如表3所示. 首先, 将分离得到的次要异构体3aa'溶于DMF中, 加入1.5 equiv. tBuOLi为碱, 在室温条件下, 大部分3aa'转化为主要异构体3aa (Table 3, Entry 1). 然而, 当使用Et3N作为碱时, 没有观察到明显的异构化反应(Table 3, Entry 2); 而使用Cs2CO3作为碱时, 仅有少量3aa'转化为3aa (Table 3, Entry 3). 此外, 当以PhCF3为溶剂时, 即使使用tBuOLi作为碱, 也没有观察到3aa'向3aa的转化(Table 3, Entry 4). 这些实验结果表明, 极性溶剂DMF能够显著促进螺环β-内酰胺混合物的retro-aldol/aldol异构化过程, 从而推动次要非对映体向主要非对映体的转化. 同时, 强碱的存在对这一过程起到了关键作用.
表3 控制实验Table 3 Control experiment |
| Entry | Base | Solvent | Conversion rateb |
|---|---|---|---|
| 1 | tBuOLi | DMF | ≈85% |
| 2 | Et3N | DMF | n.d. |
| 3 | Cs2CO3 | DMF | ≈15% |
| 4 | tBuOLi | PhCF3 | n.d. |
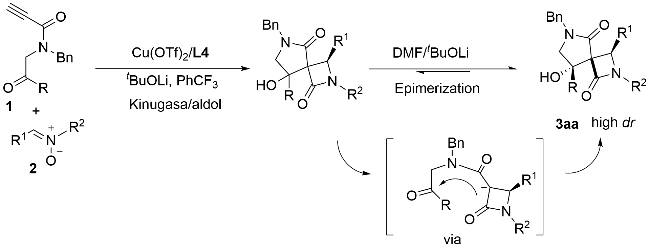
基于以上实验结果, 我们认为反应经历了两个关键步骤(图4): 首先, 在PhCF3溶剂中, 通过Kinugasa/Aldol串联反应过程, 生成了两种非对映异构体产物; 随后, 在DMF溶剂中, 通过碱促进的retro-aldol/aldol异构化过程, 次要异构体进一步向主要异构体转化, 从而获得具有高非对映选择性的产物.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
本工作通过级联Kinugasa/aldol反应, 成功实现了一类手性螺环β-内酰胺化合物的高效不对称合成. 该方法在温和的反应条件下, 利用铜催化烷基酮连接的丙炔酰胺与硝酮进行不对称Kinugasa反应, 随后通过分子内aldol反应实现螺环化. 尽管初始产物的非对映选择性(dr值)较低, 但通过在极性溶剂DMF中引入碱促进的retro-aldol/aldol过程, 能够部分地将次要异构体转化为主要异构体, 从而实现非对映体富集, 显著提高了反应的非对映选择性. 这一策略为合成结构新颖、产率良好且对映选择性优异的手性螺环β-内酰胺提供了一条高效途径. 目前, 我们正在进一步探索该方法在手性螺杂环合成中的应用, 相关研究正在进行中.
4 实验部分
在氩气氛围下, 将丙炔酰胺1 (0.20 mmol, 1.0 equiv.)、硝酮2 (0.24 mmol, 1.2 equiv.)、tBuOLi (0.3 mmol, 24 mg, 1.5 equiv.)、Cu(OTf)2 (0.02 mmol, 7.5 mg, 10 mol%)与手性配体L4 (0.024 mmol, 8 mg, 12 mol%)加入到2 mL无水PhCF3中, 并在0 ℃下搅拌12 h. 然后除去溶剂PhCF3, 将混合物置于DMF (2 mL)中在室温下继续搅拌5 h. 通过薄层色谱(TLC)监测反应完成后, 向混合物中加入H2O (5.0 mL)和乙酸乙酯(5.0 mL). 分离有机相, 用乙酸乙酯(5.0 mL×3)萃取水相. 将合并的有机相分别用H2O和饱和食盐水洗涤, Na2SO4干燥. 通过减压蒸馏除去有机溶剂, 残余物通过硅胶色谱柱(V(乙酸乙酯)/V(石油醚)=1/7~1/2)纯化, 得到所需产物3aa~3dh.
(Cheng, B.)