

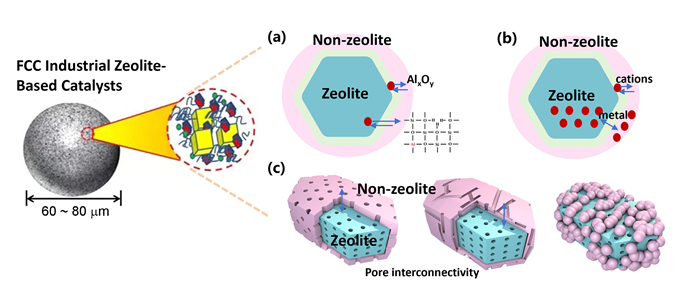
1 引言
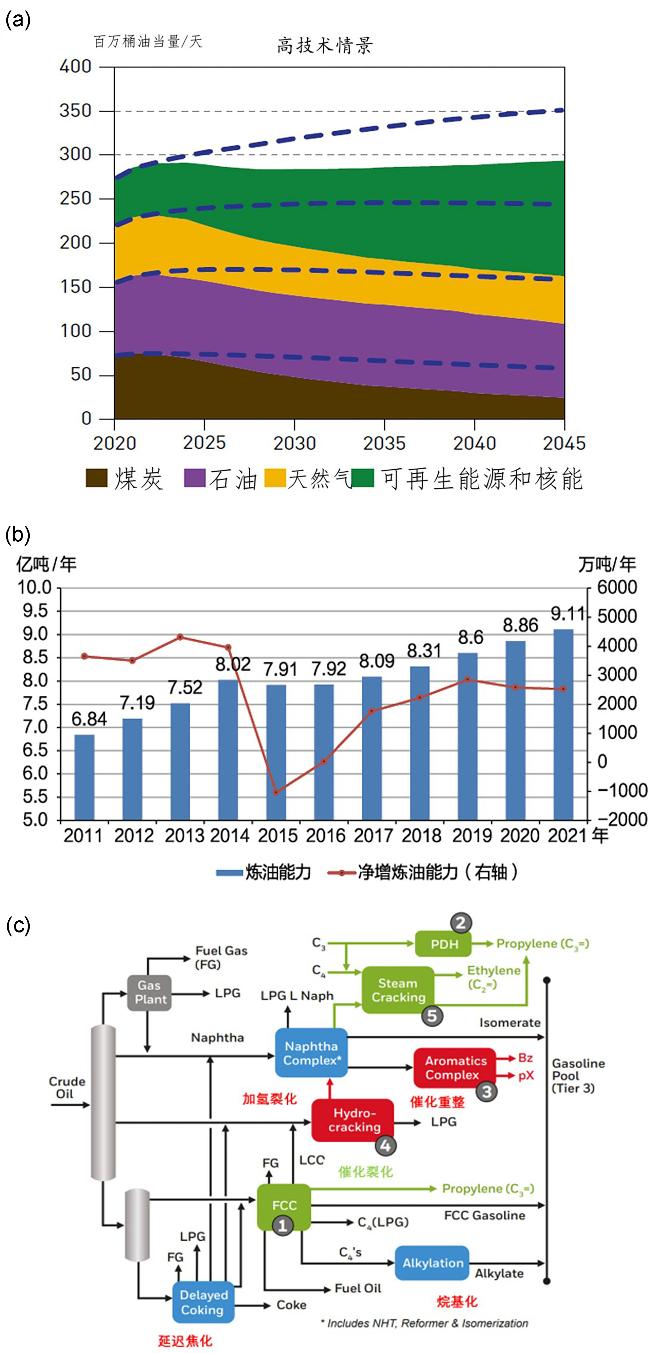
图1 炼油化工行业发展现状: (a) OPEC世界石油展望; (b)炼油能力数据表(数据统计来源于国家统计局)及(c)典型的炼油+化工工艺路线(来源于UOP).Figure 1 Current status of the refining and petrochemical industry: (a) global energy outlook by OPEC, (b) refining capacity data (sourced from the National Bureau of Statistics of China), and (c) typical integrated refining and petrochemical process flow (sourced from UOP) |
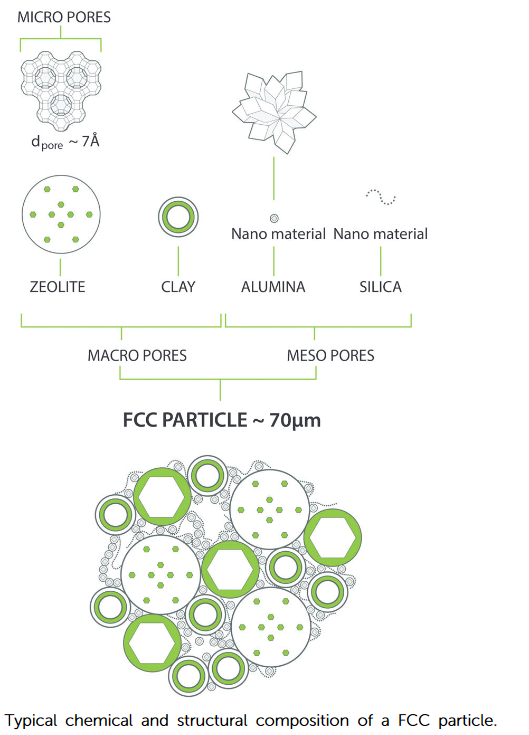
2 催化裂化多组元催化剂的等级特征
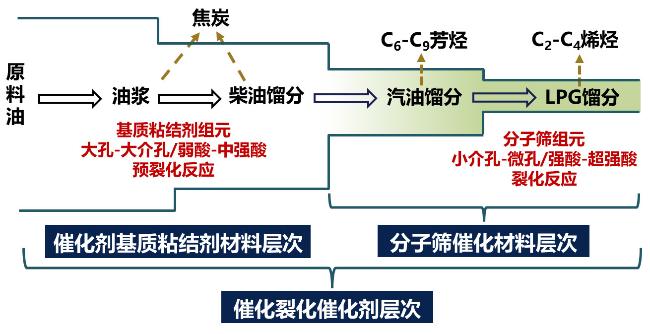
3 多组元催化剂的等级结构协同接力催化功能
4 沸石-非沸石一体化催化剂组元间的相互作用
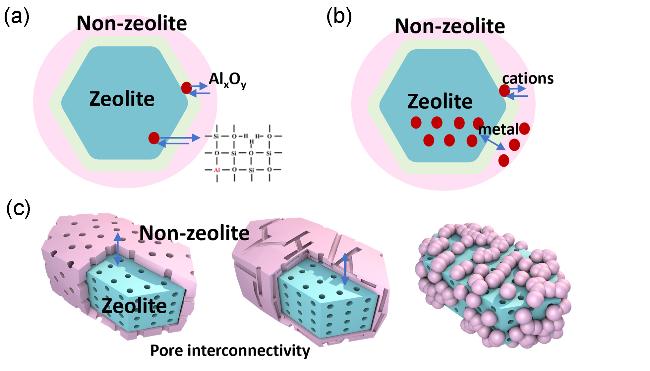
4.1 铝物种在沸石与非沸石组元之间的迁移及其导致的酸性质变化
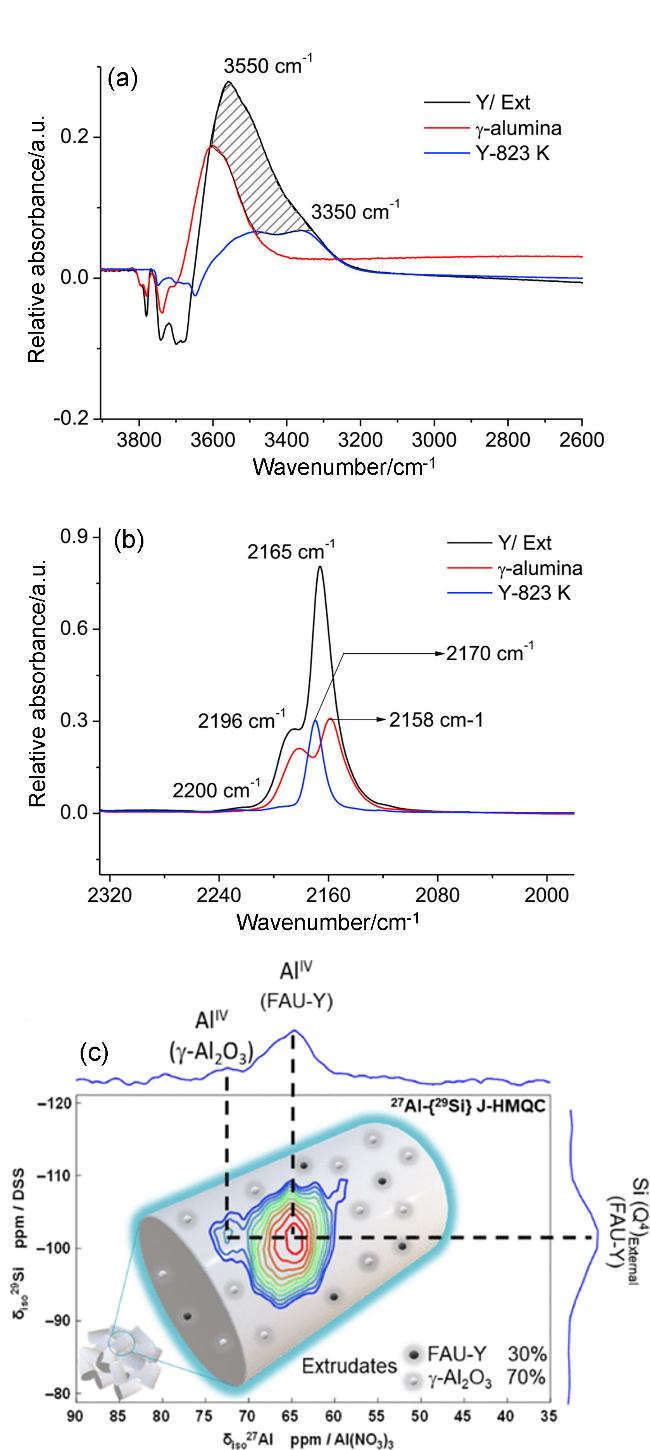
图5 组元间相互作用导致的新酸性位点的生成[42-43]: (a, b)沸石及其挤出物上CO吸附前后的差异光谱[43][(a) OH区, (b) CO区]及(c)沸石挤出物的27Al-{29Si} J-HMQC NMR谱图[42]Figure 5 Formation of new acid sites induced by intercomponent interactions[42-43]: (a, b) difference spectra before and after CO saturation on zeolite and its extrudates[43] [(a) OH region (b) CO region] (Copyright 2020, with permission from Elsevier) and (c) 27Al-{29Si} J-HMQC NMR spectra on extrudates[42] (Copyright 2021, American Chemical Society) |
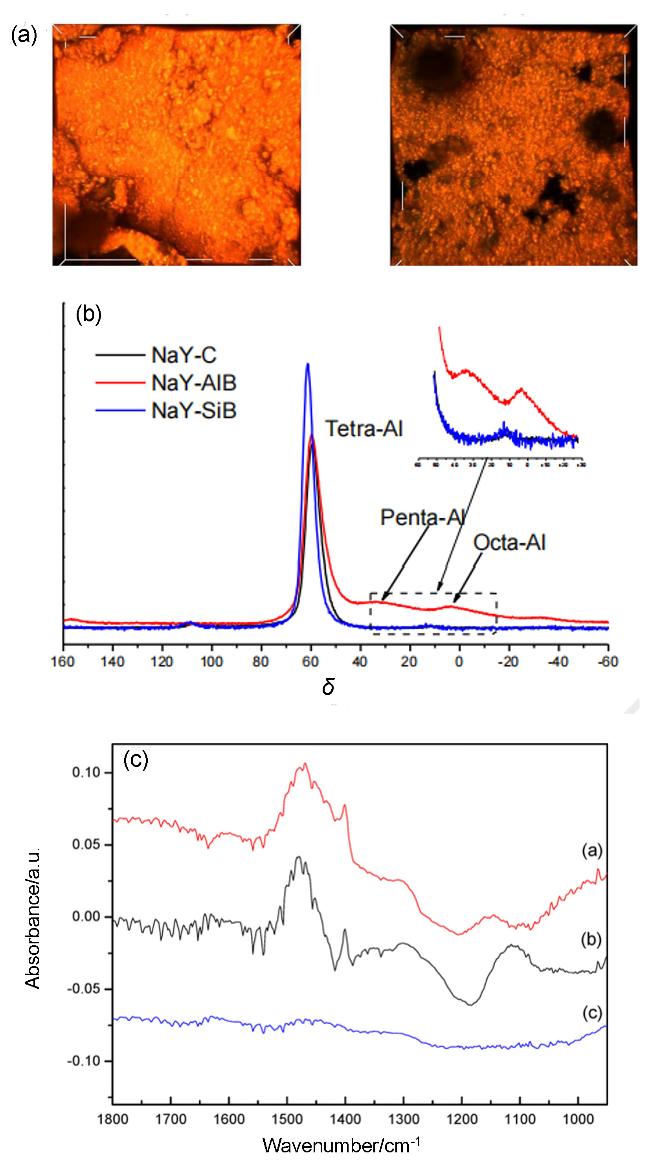
图6 二氧化硅作为粘结剂对催化剂酸性特征的影响[55-57]: (a) ZSM-5沸石及SiO2(左图)或Al2O3(右图)非沸石组元复合催化剂的三维共聚焦荧光显微镜图像[55]、(b)复合催化剂的27Al NMR谱图[56]及(c) NH3吸附的FT-IR光谱在不同多组元催化剂上(NaY-SiO2, NaY, SiO2[57])Figure 6 Effect of silica as a binder on the acidity of catalysts[55-57]: (a) 3D confocal fluorescence microscopy images of the ZSM-5-binder-bound pellets combined with either SiO2 (left) or Al2O3 (right) as non-zeolitic components[55] (Copyright 2019, Bert M. Weckhuysen, Martijn Burgers, Anton-Jan Bons, et al. Published by Wiley-VCH Verlag GmbH & Co. KGaA), (b) 27Al solid state MAS NMR of multicomponent catalysts[56] (Copyright 2018 Elsevier B. V. All right reserved), and (c) FT-IR of NH3 adsorbed on different multicomponent catalyst (NaY-SiO2, NaY, and SiO2)[57] (Copyright 2015, American Chemical Society) |
4.2 金属位点在沸石与非沸石组元之间的迁移与交换
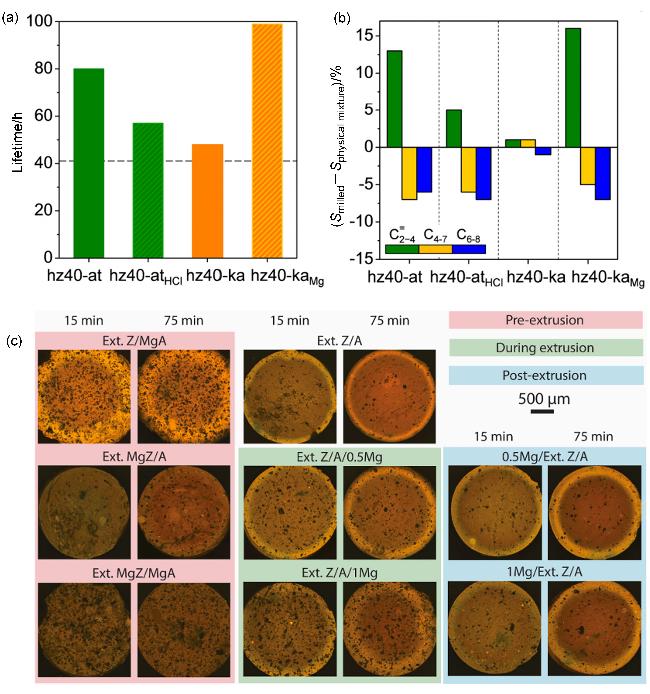
图7 金属阳离子在沸石与非沸石组元之间的迁移: (a)不同催化剂(多级孔ZSM-5与凹凸棒、HCl处理的凹凸棒土、高岭土和MgO掺杂高岭土的混合物)上的MTH反应寿命[64]、(b)对比相应物理混合催化剂的平均产物选择性的差异[64]及(c)沸石基成型催化剂早期失活阶段的可视化共聚焦荧光显微镜(CFM)图像[66]Figure 7 Migration of metal cations between zeolitic and non-zeolitic components: (a) MTH lifetime over milled hierarchical ZSM-5 admixtures with attapulgite, HCl-treated attapulgite, kaolin, and MgO-doped kaolin,[64] (b) differences in average product selectivity compared with respect to the corresponding physical mixtures,[64] (Copyright 2014, American Chemical Society) and (c) visualizing early stages of deactivation in zeolite-based shaped catalyst bodies by confocal fluorescence microscopy (CFM) images[66] (Copyright 2023 The Nikolaos Nikolopoulos, Luke A. Parker, Maurits W. Vuijk, Bert M. Weckhuysen. Published by Elsevier Inc) |
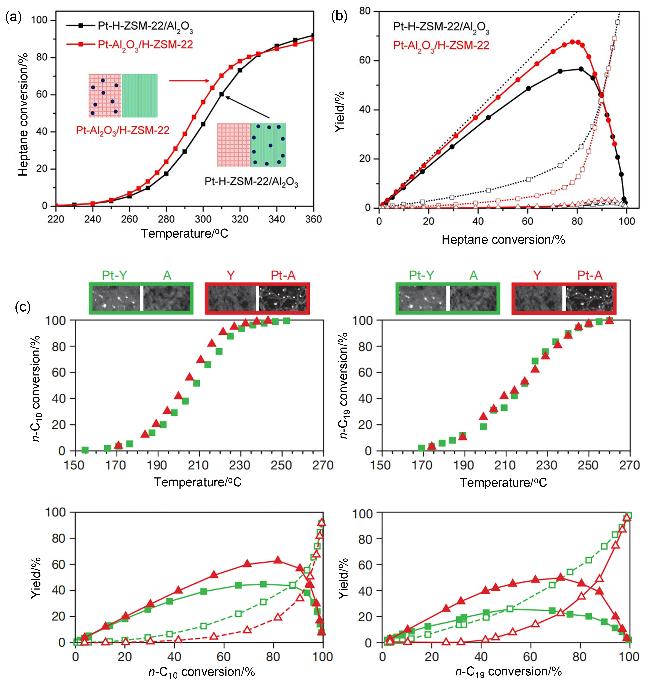
图8 金属颗粒在整体催化剂上分布位点的影响: (a)正庚烷转化率与反应温度的关系及(b)总C7异构体和裂解产物的产率[72]以及(c)组元间纳米尺度的邻近对加氢裂化活性和选择性的影响[73]Figure 8 Effect of metal particle deposition sites on the performance of composite catalysts: (a) n-heptane conversion against the reaction temperature, and (b) yields of total C7 isomers and cracking products[72] (Copyright 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim) and (c) impact of nanoscale intimacy on hydrocracking activity and selectivity[73] (Copyright 2015, Springer Nature Limited) |
4.3 沸石组元与非沸石组元的孔道匹配联通性
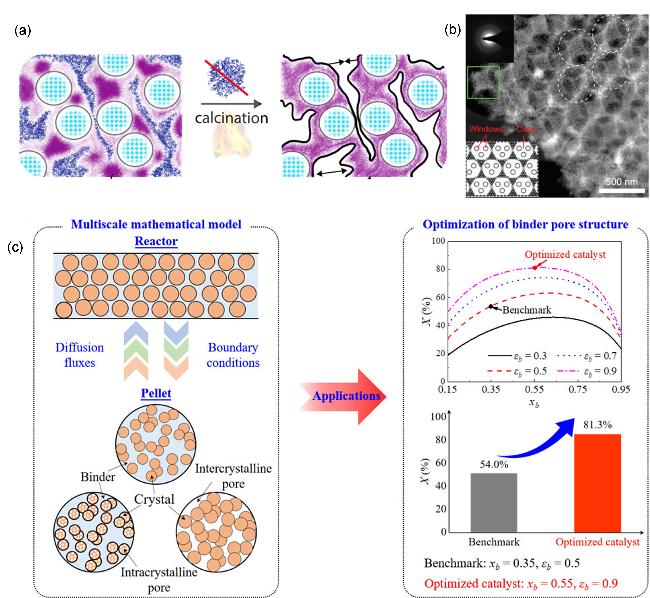
图9 非沸石组元的引入对整体催化剂的孔梯度影响: (a)非沸石组元的引入使得整体催化剂的孔梯度重排[4]、(b)构建从大孔到微孔多尺度孔径大小匹配的多孔材料, 提高质量传输[22]及(c)组元间孔径匹配方面存在强扩散限制[80]Figure 9 Effect of incorporating non-zeolitic components on the pore hierarchy of the overall catalyst: (a) pore hierarchy reorganization induced by non-zeolitic components[4] (Copyright 2019, American Chemical Society), (b) construction of multiscale pores with well-matched pore sizes from macropores to micropores enhances mass transport[22] (Copyright 2022, Sun, Ming-Hui; Gao Shu-shu 2022. Published by Oxford University Press on behalf of China Science Publishing) and (c) significant diffusion limitations arise from poor pore-size matching between components |
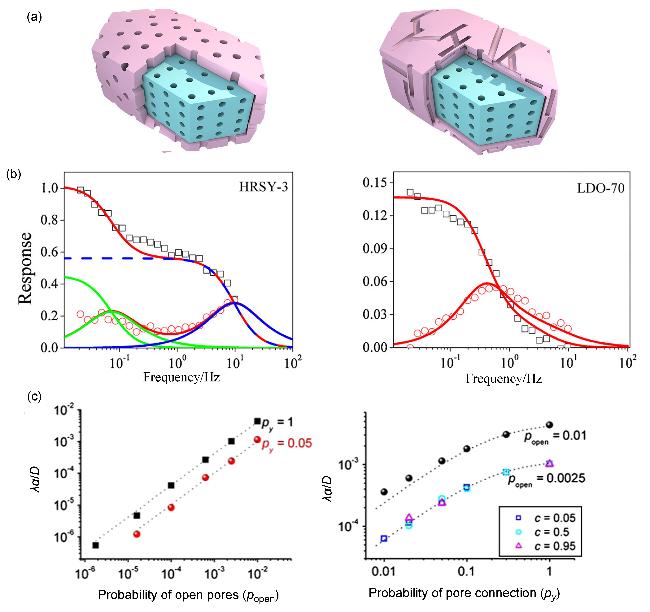
图10 (a)沸石组元与非沸石组元孔道取向引起的连通程度改变[81,84]示意图(左图为沸石组元(蓝)与非沸石组元(粉)的微介孔道取向一致, 右图为沸石组元(蓝)与非沸石组元(粉)的微介孔道取向不一致)、(b)频率相应(FR)技术对沸石-非沸石界面阻力的识别[81]及(c)由沸石晶体孔隙堵塞引起的表面传质阻力[84]Figure 10 (a) Schematic illustration of pore interconnectivity alteration induced by orientation mismatch between zeolitic and non-zeolitic components[81,84] [Left panel shows aligned orientation between the micropores of the zeolitic component (blue) and the mesopores of the non-zeolitic component (pink). Right panel shows a misaligned pore orientation between the zeolitic and non-zeolitic components], (b) frequency response (FR) technique for identifying zeolitic and non-zeolitic components interfacial resistance[81] (Copyright © 2014 Published by Elsevier B.V. All rights reserved), and (c) surface mass transfer resistance caused by blocked pore openings on zeolite crystals[84] (Copyright 2011 Aptara Inc. All rights reserved) |
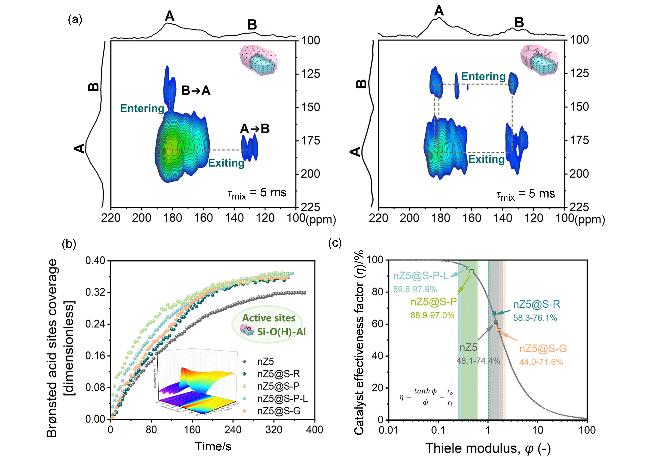
图11 沸石-非沸石组元模型催化剂孔道匹配联通性对扩散及反应动力学的影响[34]: (a) HP 129Xe NMR对组元间扩散行为的识别、(b)时间分辨原位红外光谱对多组元扩散系数的测试及(c)催化效率因子的评估Figure 11 Crucial roles of pore interconnectivity between zeolitic and non-zeolitic components in enhancing diffusion and catalytic efficiency[34]: (a) Identification of multicomponent hierarchical pore network connectivity by HP 129Xe NMR, (b) relating pore interconnectivity to diffusion in multicomponent model catalysts by time-resolved rapid-scan in situ FTIR technology, and (c) catalytic efficiency assessment of multicomponent model catalysts (Copyright 2025, American Chemical Society) |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
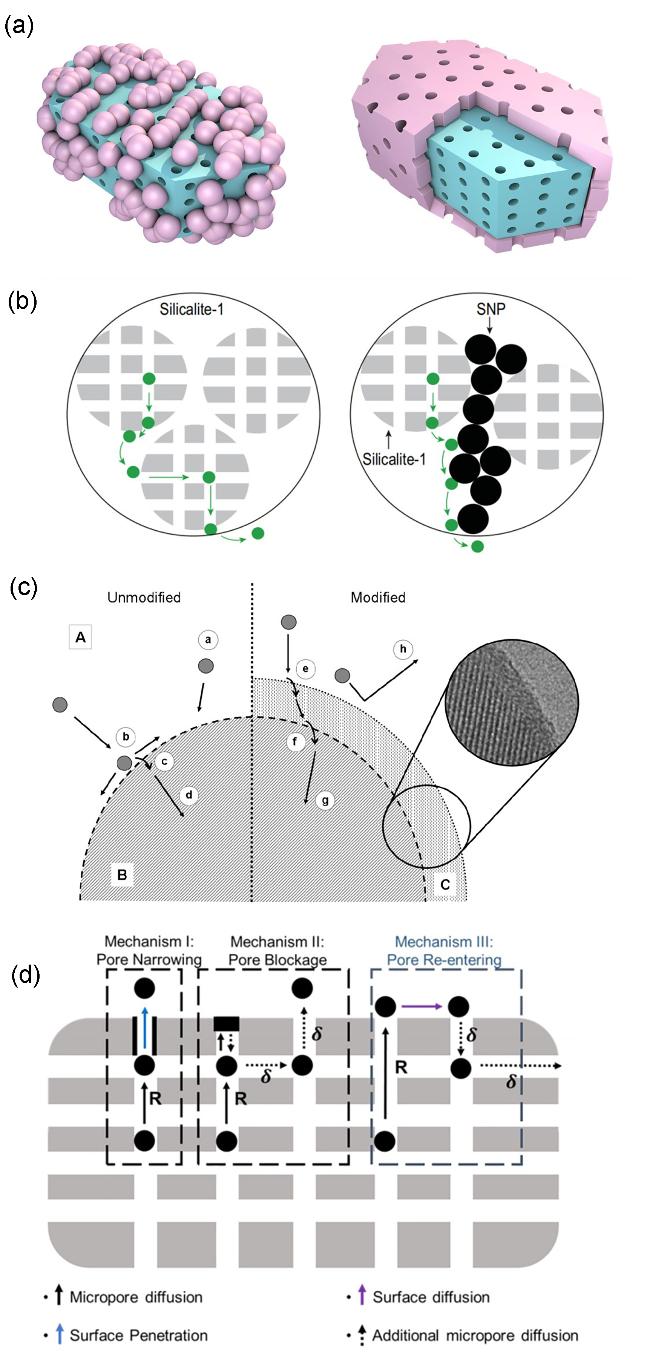
图12 (a)沸石组元与非沸石组元空间相对位置分布导致的扩散路径改变示意图[75-76,95](左图为非沸石组元的填充较为松散的情况; 右图为非沸石组元的填充相对较为紧密的情况)、(b)客体分子在纳米沸石中的扩散路径以及表面覆盖硅球的影响[75]及(c, d)沸石组元表面质量传输[76,95]Figure 12 (a) Schematic of the effect of the spatial distribution between zeolitic and non-zeolitic components on diffusion pathways[75-76,95] (The left panel illustrates a loosely packed configuration of non-zeolitic components, whereas the right panel shows more uniformly coated), (b) guest molecules in nano-zeolite and the impact of surface covering with small silica particles[75]. (Copyright 2019, Zhou, Jian; Fan, Wei 2019. Published by Oxford University Press on behalf of China Science Publishing) and (c, d) surface barrier schemes for mass transport in zeolitic component[76,95] (Copyright 2021, Shen Hu, Junru Liu, Guanghua Ye, et al. Angewandte Chemie International Edition published by Wiley VCH GmbH. Copyright 2011, American Chemical Society) |