


1 引言
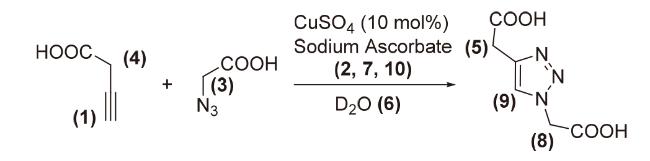
2 结果与讨论
2.1 CuAAC反应过程中转动扩散系数减小
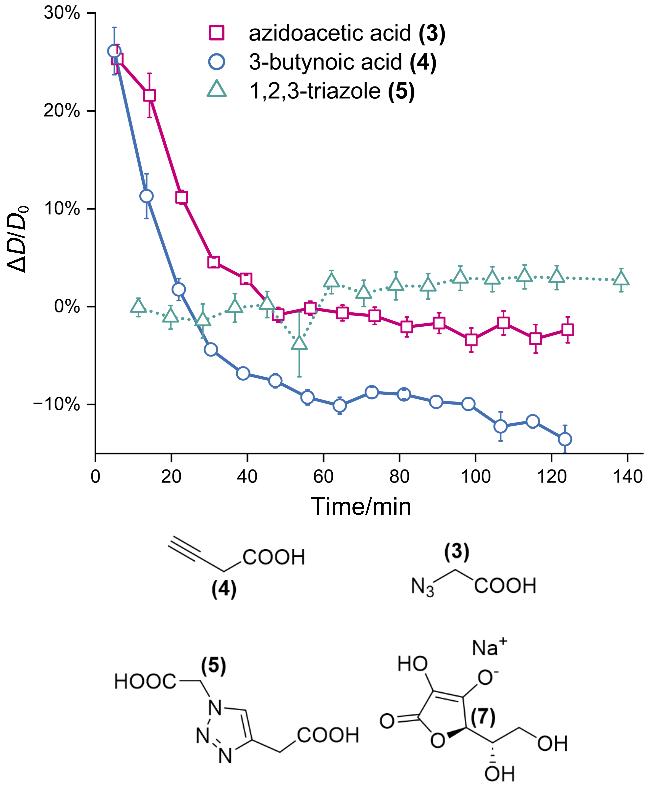
图2 CuAAC反应过程中平动扩散系数随时间的变化. 数字对应于拟合扩散系数所用的核磁1H信号, 3-丁炔酸与叠氮乙酸的起始浓度为230 mmol/L, CuSO4浓度为10 mol%, 抗坏血酸钠浓度为40 mol%Figure 2 Translational diffusion coefficients plotted as a function of time during the CuAAC reaction. Numbers correspond to the 1H signal used to fit the translational diffusion coefficient, the starting concentration of 3-butynoic acid and azidoacetic acid were both 230 mmol/L, the concentration of CuSO4 was 10 mol% of the reactant, and the concentration of sodium ascorbate was 40 mol% of the reactant |
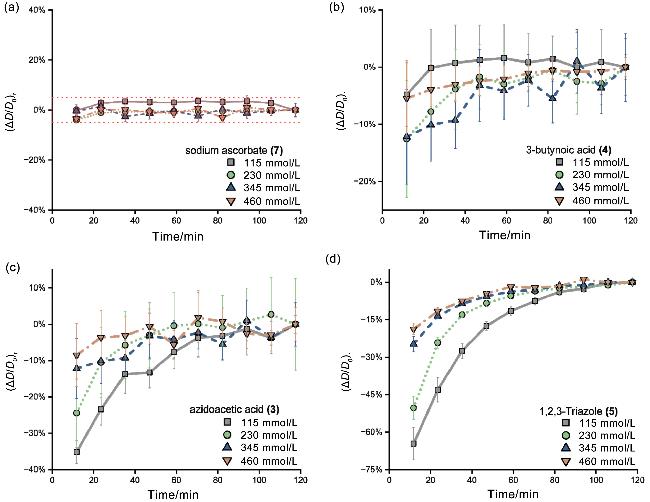
图3 不同浓度下CuAAC反应过程中转动扩散系数随时间的变化. 图中浓度为反应物起始浓度. (a)基于抗坏血酸钠的(7)信号拟合的转动扩散系数随时间的变化; (b)基于3-丁炔酸的(4)信号拟合的转动扩散系数随时间的变化; (c)基于叠氮乙酸的(3)信号拟合的转动扩散系数随时间的变化; (d)基于三氮唑的(5)信号拟合的转动扩散系数随时间的变化.Figure 3 Rotational diffusion coefficient over time during CuAAC reaction at different concentrations. The concentrations in the figure are the starting concentrations of the reactants. (a) Rotational diffusion coefficients of sodium ascorbate over time, fitted with the NMR signals of (7); (b) rotational diffusion coefficients of 3-butynoic acid over time, fitted with the NMR signals of (4); (c) rotational diffusion coefficients of azidoacetic acid plotted as a function of time, fitted with the NMR signals of (3); (d) rotational diffusion coefficients plotted as a function of time of 1,2,3-triazole, fitted with the NMR signals of (5) |
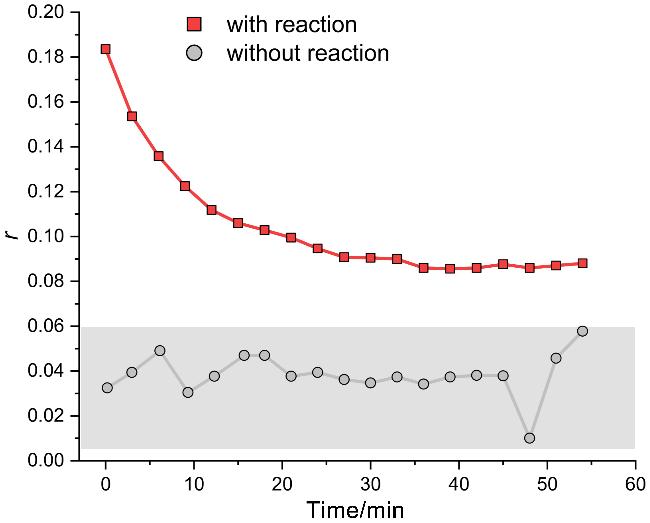
2.2 反应过程中非经典转动扩散现象原因的讨论
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
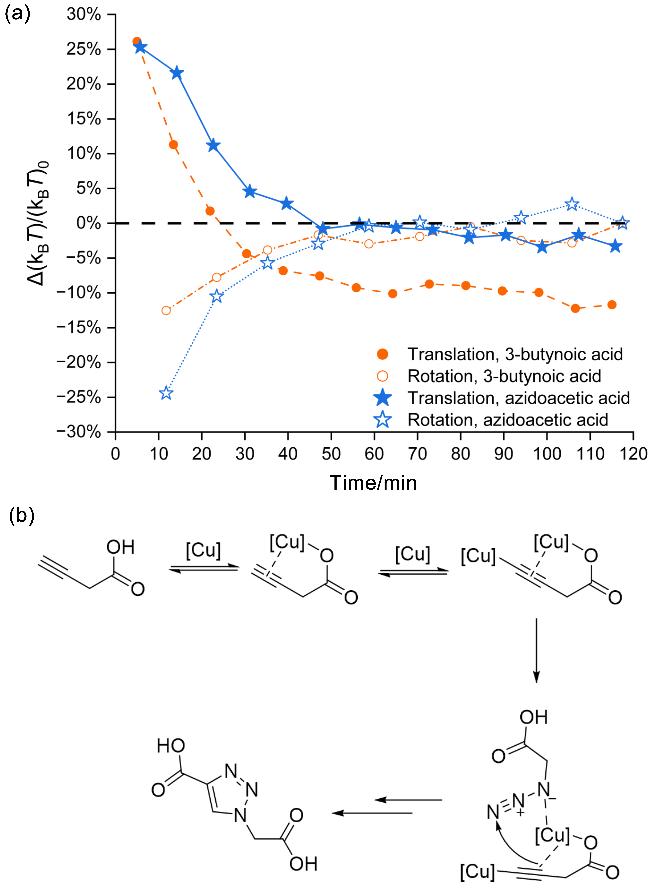
图5 平动能量和转动能量变化的可能机制. (a)反应过程中3-丁炔酸和叠氮乙酸平动和转动能量改变幅度随时间的变化; (b)可能的反应机理和中间体[17b]Figure 5 Possible mechanisms for the translational and rotational energy interconversion. (a) Change of translational and rotational energy ploted as a function of time during the reaction for 3-butynoic acid and azidoacetic acid; (b) proposed reaction mechanisms and reaction intermediates.[17b] |