

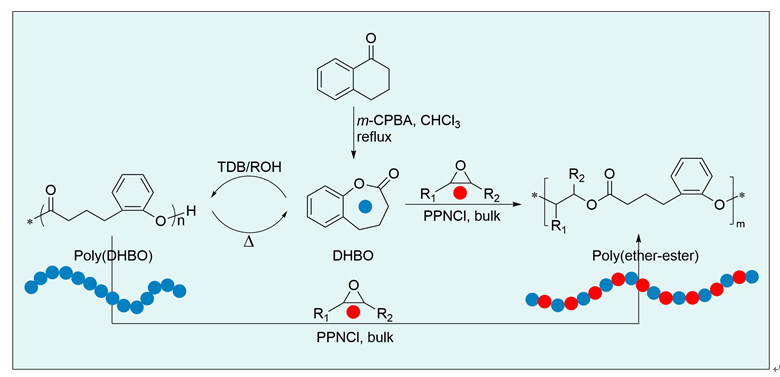
1 引言
2 结果与讨论
2.1 单体合成

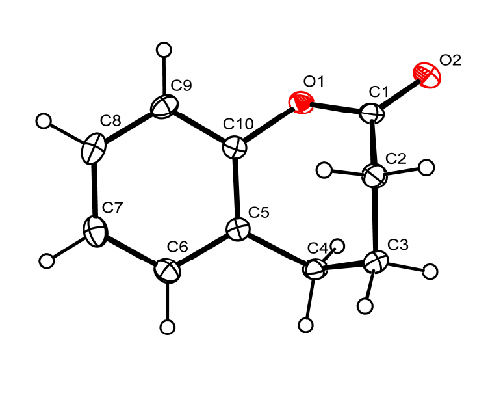
图2 单体DHBO的单晶结构Figure 2 X-ray crystal structure of DHBO Thermal ellipsoids are drawn at the 30% probability level. Selected bond lengths (nm) and angles (°): C(1)—O(1) 0.1364(2), C(1)—O(2) 0.11993(19), C(10)—O(1) 0.14052(18), O(1)—C(1)—O(2) 117.57(14), C(2)—C(1)—O(2) 117.29(13), C(1)—O(1)—C(10) 120.60(11) |
2.2 DHBO的开环均聚合
表1 TBD催化DHBO的均聚合aTable 1 Homopolymerization of DHBO mediated with TBD |
| Entry | [M]0/[Cat.]/[ROH] | Solvent | [M]0/(mol•L-1) | t/h | T/℃ | Conv.b/% | Mn,GPCc/(kg•mol-1) | Ðc |
|---|---|---|---|---|---|---|---|---|
| 1 | 100/1/1 | toluene | 1 | 5 | r.t. | 73 | 4.79 | 1.88 |
| 2 | 100/1/1 | toluene | 2 | 5 | r.t. | 85 | 7.17 | 1.86 |
| 3 | 100/1/1 | toluene | 4 | 5 | r.t. | 93 | 6.06 | 1.62 |
| 4 | 100/1/1 | THF | 2 | 5 | r.t. | 77 | 6.03 | 1.62 |
| 5 | 100/1/1 | DMF | 2 | 5 | r.t. | 36 | 1.36 | 1.89 |
| 6 | 100/1/1 | DCM | 2 | 5 | r.t. | 48 | 3.00 | 1.24 |
| 7 | 100/1/1 | DCM | 2 | 24 | r.t. | 55 | 8.75 | 2.15 |
| 8 | 100/1/1 | — | — | 10 min | 90 | 90 | 7.49 | 2.09 |
| 9 | 100/1/1 | — | — | 10 min | 110 | 92 | 6.21 | 1.93 |
| 10 | 300/1/1 | — | — | 0.5 | 90 | 84 | 9.00 | 1.89 |
| 11 | 500/1/1 | — | — | 0.5 | 90 | 80 | 10.20 | 2.00 |
a Conditions: ROH=4-MeBnOH, reactions quenched with benzoic acid, Cat: 10 μmol; b Conversion determined by 1H NMR spectroscopy; c Molecular weight (Mn) and Ð were determined by gel permeation chromatography (GPC) in DMF. |
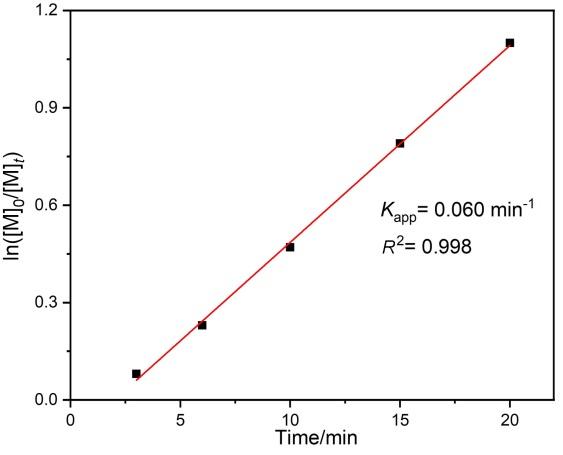
2.3 均聚物poly(DHBO)热学性质表征
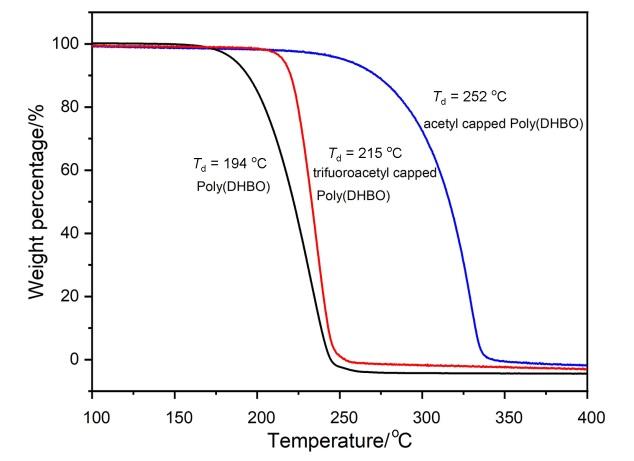
2.4 均聚物poly(DHBO)的化学回收
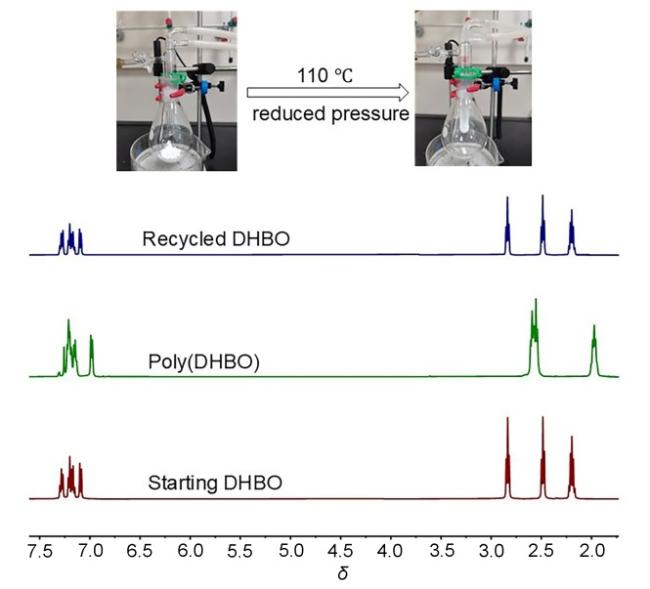
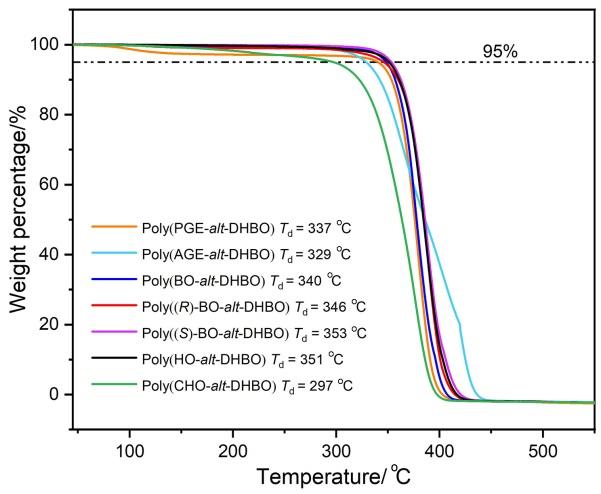
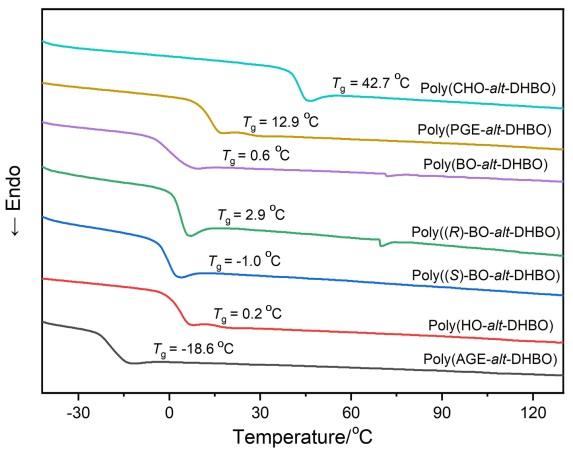
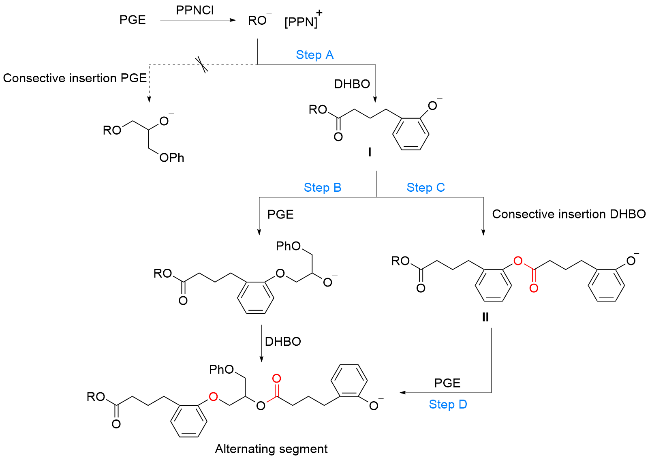
2.5 DHBO与环氧化合物的共聚合
表2 PPNCl催化DHBO和环氧化合物的共聚合aTable 2 Copolymerization of DHBO with epoxides using PPNCl 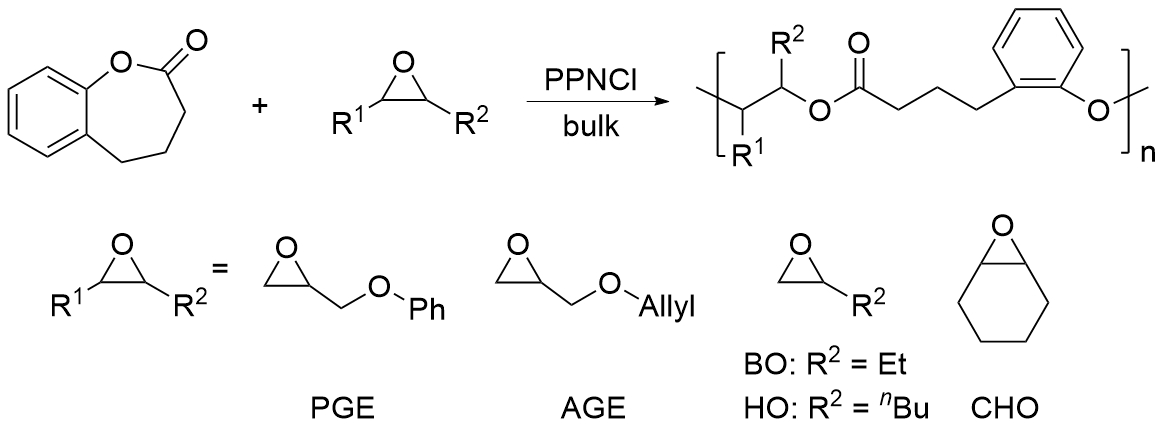 |
| Entry | [Epoxide]0/[M]0/[PPNCl]0 | Epoxide | T/℃ | Conv.b/% | Alternatingc/% | Regioselectivityc/% | Mnd/kDa | Ðd |
|---|---|---|---|---|---|---|---|---|
| 1 | —/100/1 | — | 110 | 0 | — | — | ||
| 2 | 20/—/1 | PGE | 110 | 16 | — | — | ||
| 3 | 200/100/1 | PGE | 110 | >99 | 98 | 99 | 14.4 | 1.45 |
| 4 | 200/100/1 | PGE | 90 | 95 | 98 | 98 | 15.0 | 1.47 |
| 5 | 200/100/1 | PGE | 60 | 75 | 96 | 94 | 4.75 | 1.31 |
| 6 | 200/100/1 | AGE | 110 | >99 | 96 | 97 | 16.1 | 1.38 |
| 7 | 200/100/1 | BO | 110 | >99 | 97 | 97 | 13.0 | 1.37 |
| 8e | 200/100/1 | R-BO | 110 | >99 | 98 | 97 | 8.9 | 1.67 |
| 9e | 200/100/1 | S-BO | 110 | >99 | 98 | 97 | 6.6 | 1.62 |
| 10 | 200/100/1 | HO | 110 | >99 | 96 | 98 | 8.8 | 1.34 |
| 11 | 200/100/1 | CHO | 110 | >99 | 95 | — | 11.3 | 1.39 |
a Reactions quenched with benzoic acid (0.12 mol/L in THF), polymerization time: 5 h, PPNCl: 10 μmol; b Conversion of DHBO, determined by 1H NMR spectroscopy; c Determined by 1H NMR spectrum of polymer; d Molecular weight (Mn) and Đ were obtained with gel permeation chromatography (GPC) in THF. e polymerization time: 8 h. |
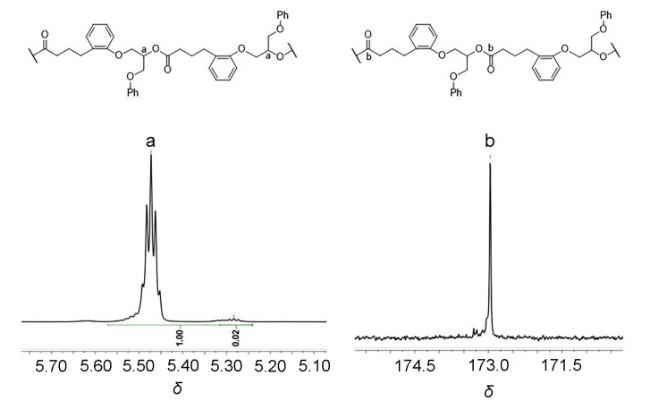

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}