

1 引言
2 结果与讨论
2.1 过渡金属氧化物为催化剂对机械合成尿素的影响
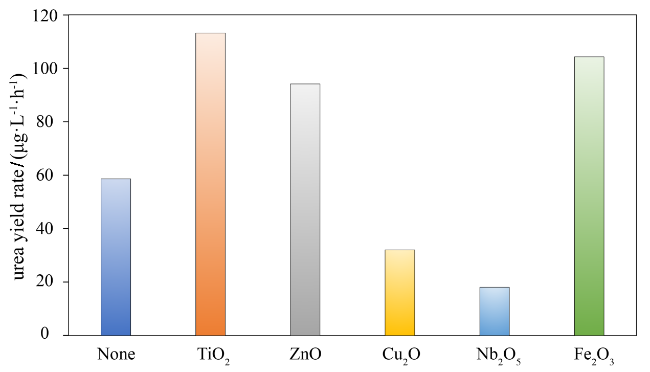
图1 加入0.5 g不同的过渡金属催化剂后机械球磨合成尿素的产率. 标为“None”的表示不加入催化剂的条件下进行机械化学合成尿素. 使用ZrO2材质的磨球和球磨罐, 磨球装载量为140 g, 转速为650 r/min, 研磨时间为1.0 hFigure 1 The mechanochemical urea yield rate with the addition of 0.5 g different metal oxide catalysts. The label “None” means performing mechanochemical urea synthesis without any additional catalysts. The grinding balls and grinding jar made of ZrO2 material were used, with a ball loading of 140 g, a rotation speed of 650 r/min, and a grinding time of 1.0 h |
2.2 N2、CO2和H2O体系TiO2机械催化合成尿素的影响因素
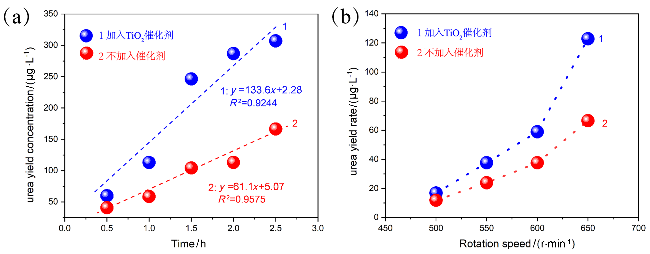
图2 N2、CO2和H2O体系机械化学合成尿素性能研究(100 mL ZrO2球磨罐和140 g磨球). (a)机械球磨尿素产量和球磨时间的关系; (b)机械球磨尿素产率和球磨转速的关系Figure 2 The performance of mechanochemical synthesis of urea from N2, CO2 and H2O (100 mL ZrO2 ball milling jar and 140 g grinding balls). (a) Relationship between urea yield and ball milling time; (b) Relationship between urea yield and ball milling speed |
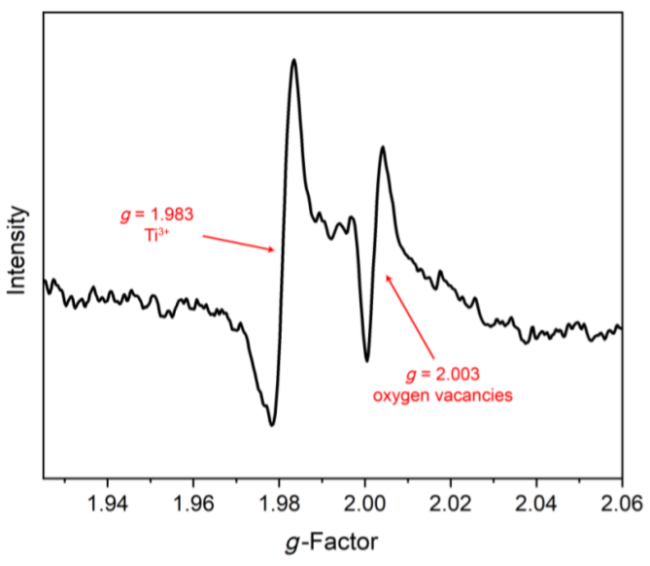
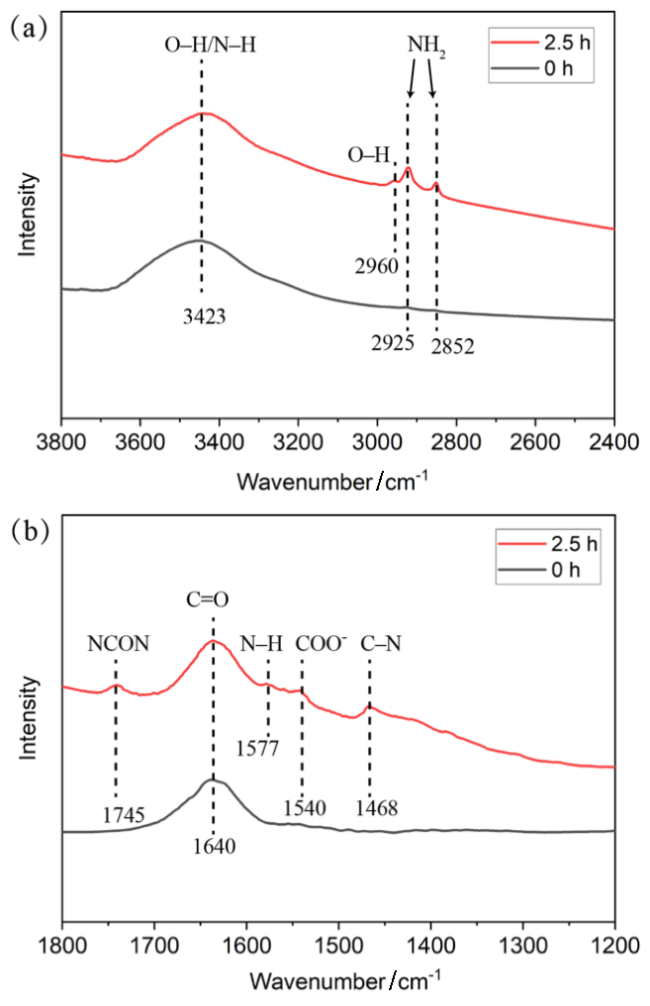
2.3 TiO2催化剂的机械催化合成尿素分析

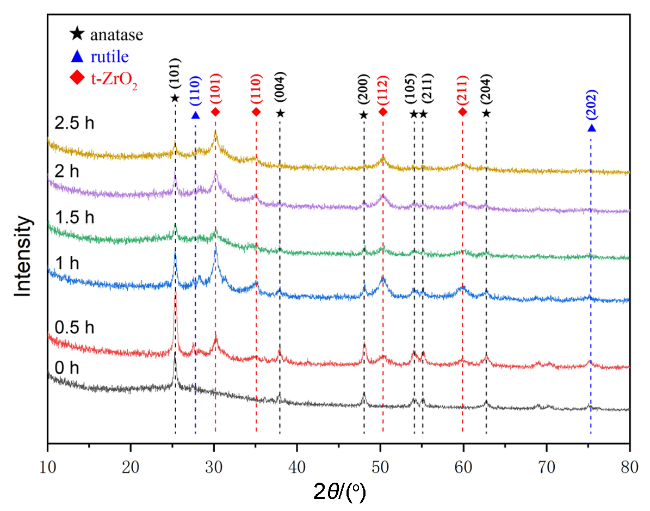
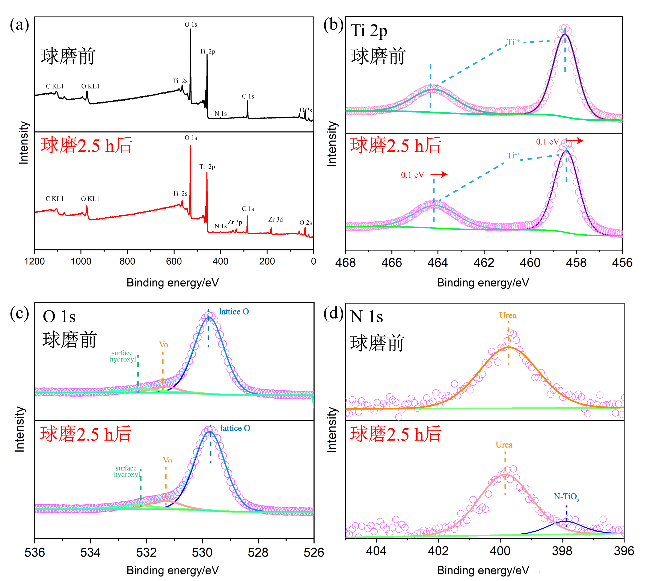
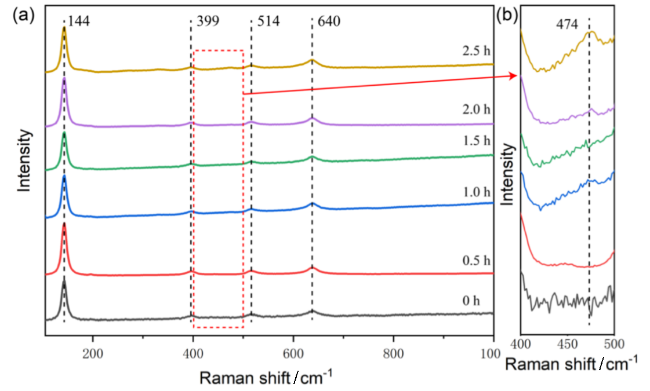
图6 机械球磨合成尿素0, 0.5, 1.0, 1.5, 2.0, 2.5 h TiO2的(a)拉曼光谱和(b) 400 cm-1到500 cm-1处的局部放大图(650 r/min, 70 g磨球)Figure 6 (a) Raman spectra and (b) local magnification in the range of 400 cm-1 to 500 cm-1 of solid powders obtained after mechanical ball milling synthesis of urea for 0, 0.5, 1.0, 1.5, 2.0, and 2.5 h (650 r/min, 70 g grinding balls) |
2.4 TiO2机械催化合成尿素的可能机理
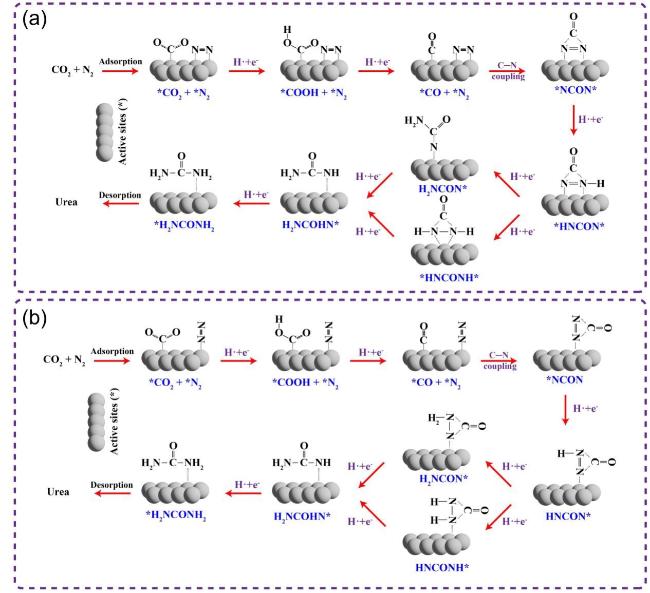
图9 通过CO2和N2机械化学合成尿素的不同机理及其中间物. 从CO2生成*CO的途径, N2以(a)侧向吸附或(b)端向吸附[52]. 路径中的H•和e-代表机械能诱导的活性氢与局域电子, 由H2O裂解及缺陷位点提供, 非电化学过程Figure 9 Different mechanisms of mechanochemical urea synthesis from CO2 and N2 and corresponding intermediate: the formation pathway of *CO from CO2, the adsorption way of N2 via (a) side-on and (b) end-on[52]. The H• and e- present in the pathway denote mechanically induced active hydrogen species and localized electrons, originating from water (H2O) splitting and defect sites, respectively; this mechanism operates independently of electrochemical processes |
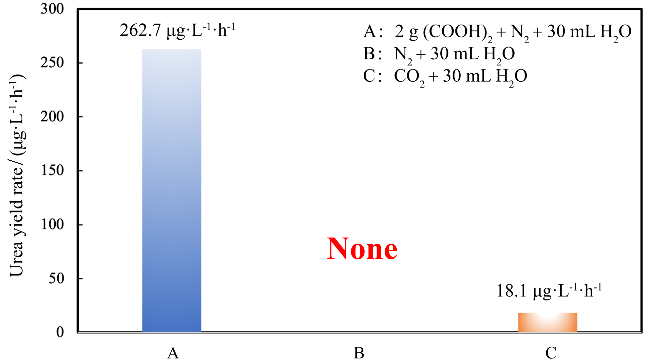
图10 分别使用A: 2 g (COOH)2, N2和30 mL H2O以及B: N2和30 mL H2O以及C: CO2和30 mL H2O为原料机械球磨合成尿素(球磨时间1.0 h, 650 r/min, 140 g磨球)Figure 10 Control experiments of mechanical ball mill urea synthesis. Urea synthesized via A: 2 g (COOH)2, N2, with 30 mL H2O, B: N2, with 30 mL H2O, and C: CO2, with 30 mL H2O (1.0 h ball milling time, 650 r/min, 140 g milling balls) |
2.5 TiO2机械催化N2+CO2+H2O体系合成尿素的密度泛函理论计算
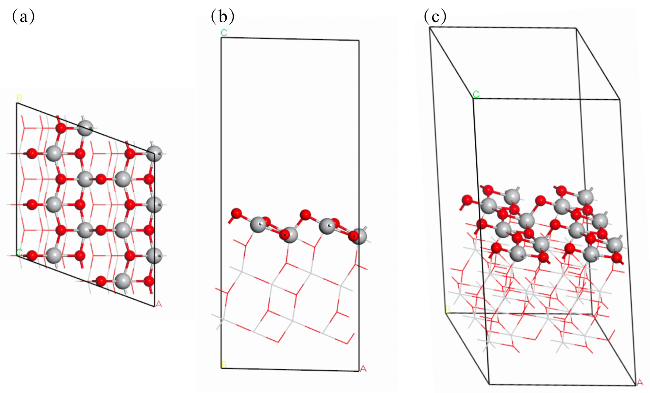
2.5.1 TiO2催化剂在机械球磨过程中氧空位(Vo)的形成机制

2.5.2 H2O在Vo-TiO2 (101)表面的化学吸附和解离
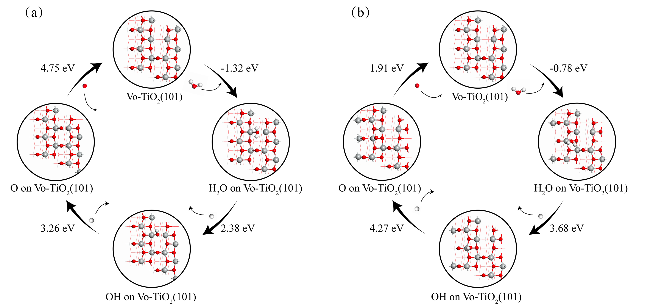
图12 H2O在Vo-TiO<H2O在Vo-TiO2 (101)表面的化学吸附和解离(每一步的形成能在图中已标出). (a)邻近Vo的Ti位点, (b)远离Vo的Ti位点(灰色小球代表Ti, 红色小球代表O, 白色小球代表H)Figure 12 The pathway of H2O molecules chemisorbed and dissociated on the Vo-TiO2 (101) surface (Each step has its corresponding formation energy value marked). (a) Ti sites adjacent to Vo, and (b) Ti sites away from Vo (grey balls represent Ti, red balls O, white balls H) |
2.5.3 N2在Vo-TiO2 (101)表面上不同吸附模式
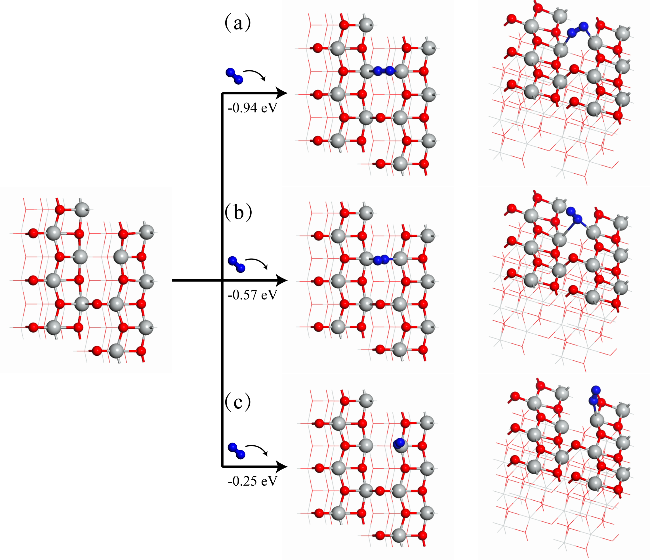
图13 几何优化后N2吸附在富含氧空位的TiO2 (101)表面上不同吸附构型: (a)在Vo上的侧向吸附方式和(b)端向吸附方式, (c)邻近Vo的端向吸附方式(对应吸附构型的吸附能已在图中标出, 灰色小球代表Ti, 红色小球代表O, 蓝色小球代表N)Figure 13 The adsorption structu The adsorption structure of N2 molecules on the TiO2 (101) surface with the presence of oxygen vacancies (Vo) after geometry optimization, with different adsorption sites: (a) side-on adsorption and (b) end-on adsorption at Vo site, and (c) end-on adsorption adjacent to Vo (The adsorption energies corresponding to these configurations are marked in the figure, grey balls represent Ti, red balls O, blue balls N) |
2.5.4 尿素在Vo-TiO2 (101)表面的生成
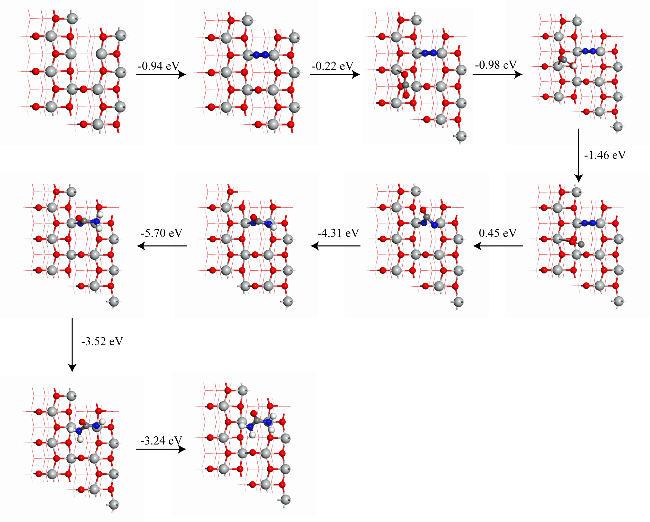
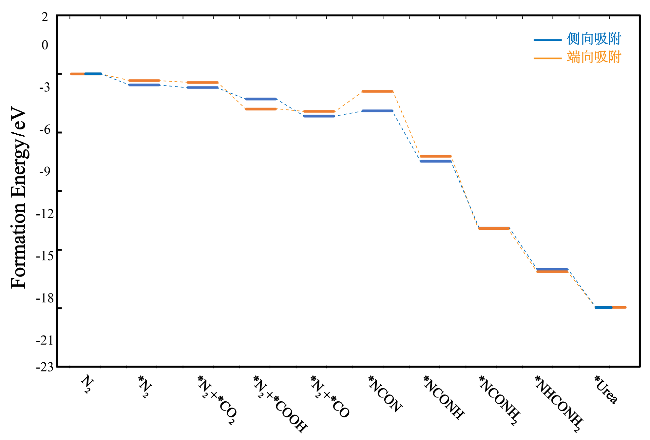
图14 N2在Vo-TiO2 (101)表面于Vo上侧向吸附时机械化学合成尿素可能的演变路线(灰色小球代表Ti, 红色小球代表O, 蓝色小球代表N, 白色小球代表H, 深灰色小球代表C)Figure 14 The possible evolution pathway for the mechanochemical synthesis of urea with the side-on adsorption of N2 on the Vo site of the Vo-TiO2 (101) surface (grey balls represent Ti, red balls O, blue balls N, white balls H, dark-grey balls C) |
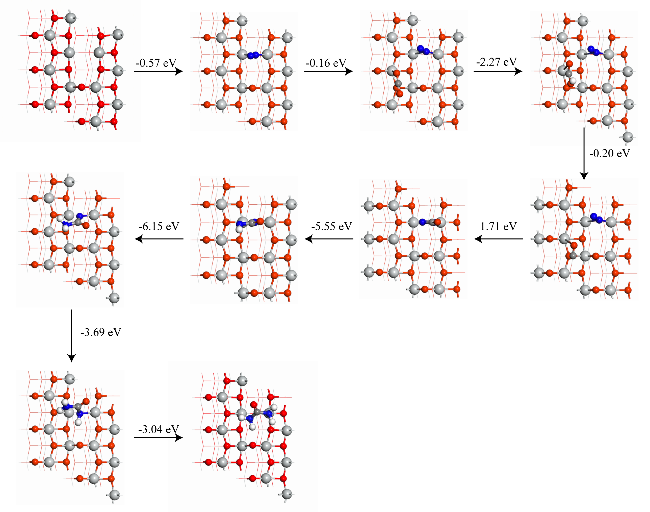
图15 N2在Vo-TiO2 (101)表面于Vo上端向吸附时机械化学合成尿素可能的演变路线(灰色小球代表Ti, 红色小球代表O, 蓝色小球代表N, 白色小球代表H, 深灰色小球代表C)Figure 15 The possible evolution pathway for the mechanochemical synthesis of urea with the end-on adsorption of N2 on the Vo site of the Vo-TiO2 (101) surface (grey balls represent Ti, red balls O, blue balls N, white balls H, dark-grey balls C) |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}