Chinese Journal of Organic Chemistry ›› 2026, Vol. 46 ›› Issue (2): 507-514.DOI: 10.6023/cjoc202507040 Previous Articles Next Articles
ARTICLES
陈梓桐a, 王力为a,*( ), 沈晓b, 戚孝天a,*()
), 沈晓b, 戚孝天a,*()
收稿日期:2025-07-29
修回日期:2025-10-03
发布日期:2025-11-05
通讯作者:
王力为, 戚孝天
基金资助:
Zitong Chena, Liwei Wanga,*(), Xiao Shenb, Xiaotian Qia,*()
Received:2025-07-29
Revised:2025-10-03
Published:2025-11-05
Contact:
Liwei Wang, Xiaotian Qi
Supported by:Share
Zitong Chen, Liwei Wang, Xiao Shen, Xiaotian Qi. A Benchmark Study of Density Functional Theory (DFT) Methods for Mo-Catalyzed Carbonyl Oxidative Addition[J]. Chinese Journal of Organic Chemistry, 2026, 46(2): 507-514.
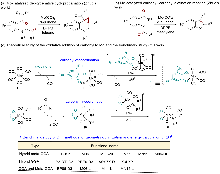
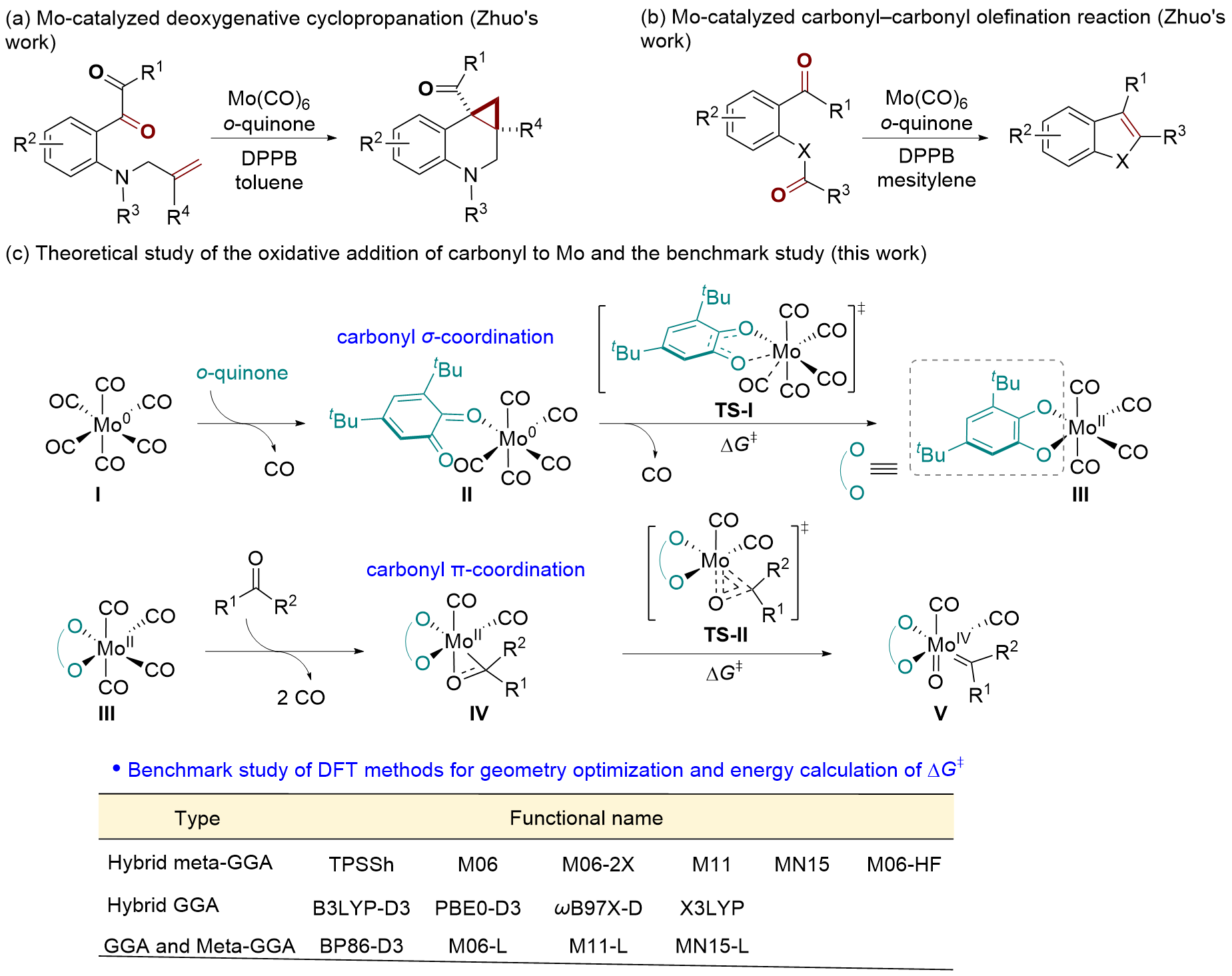
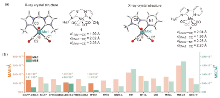
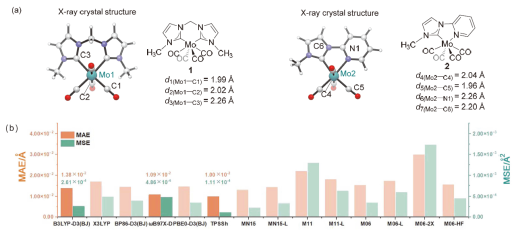

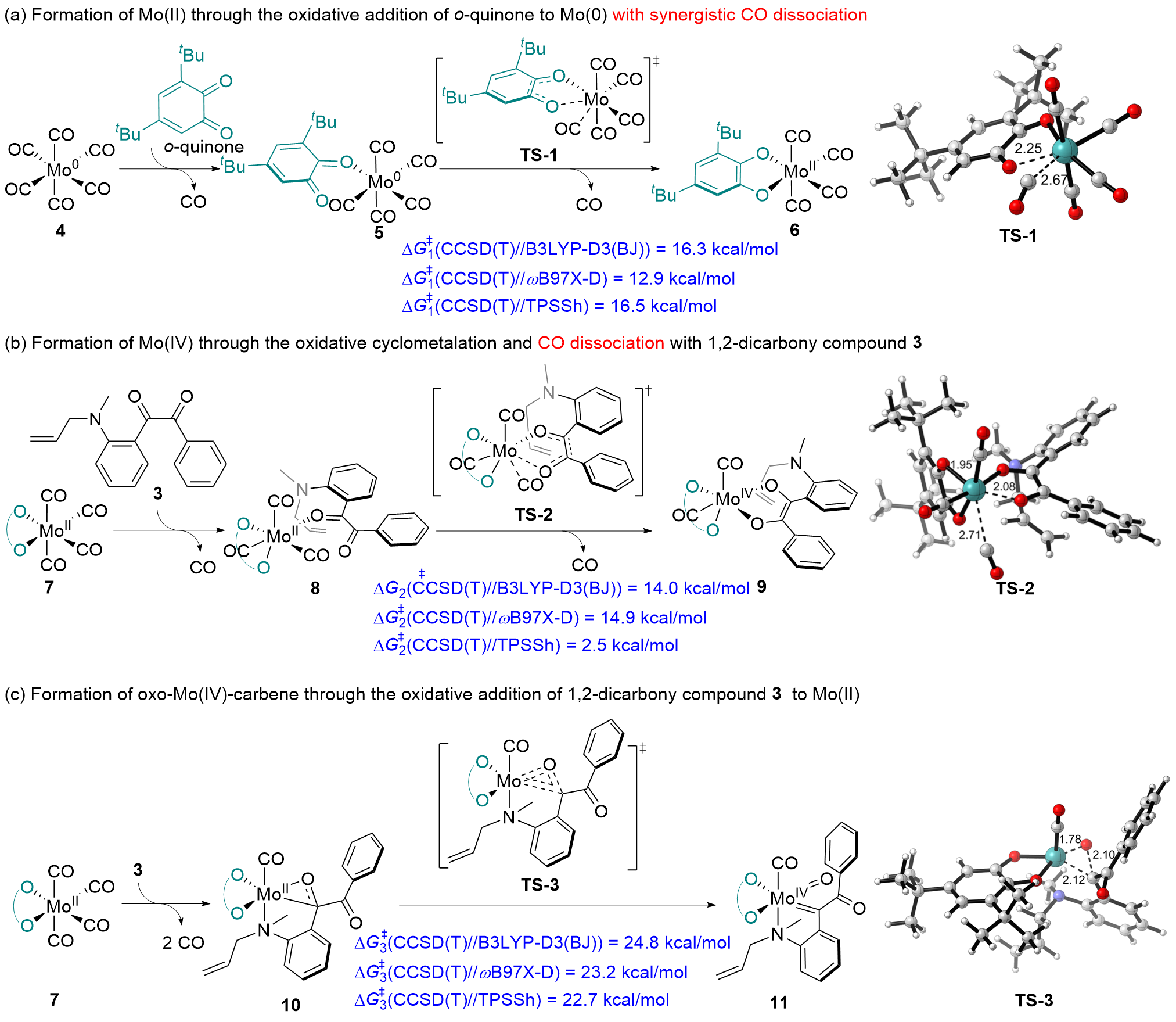
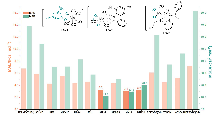
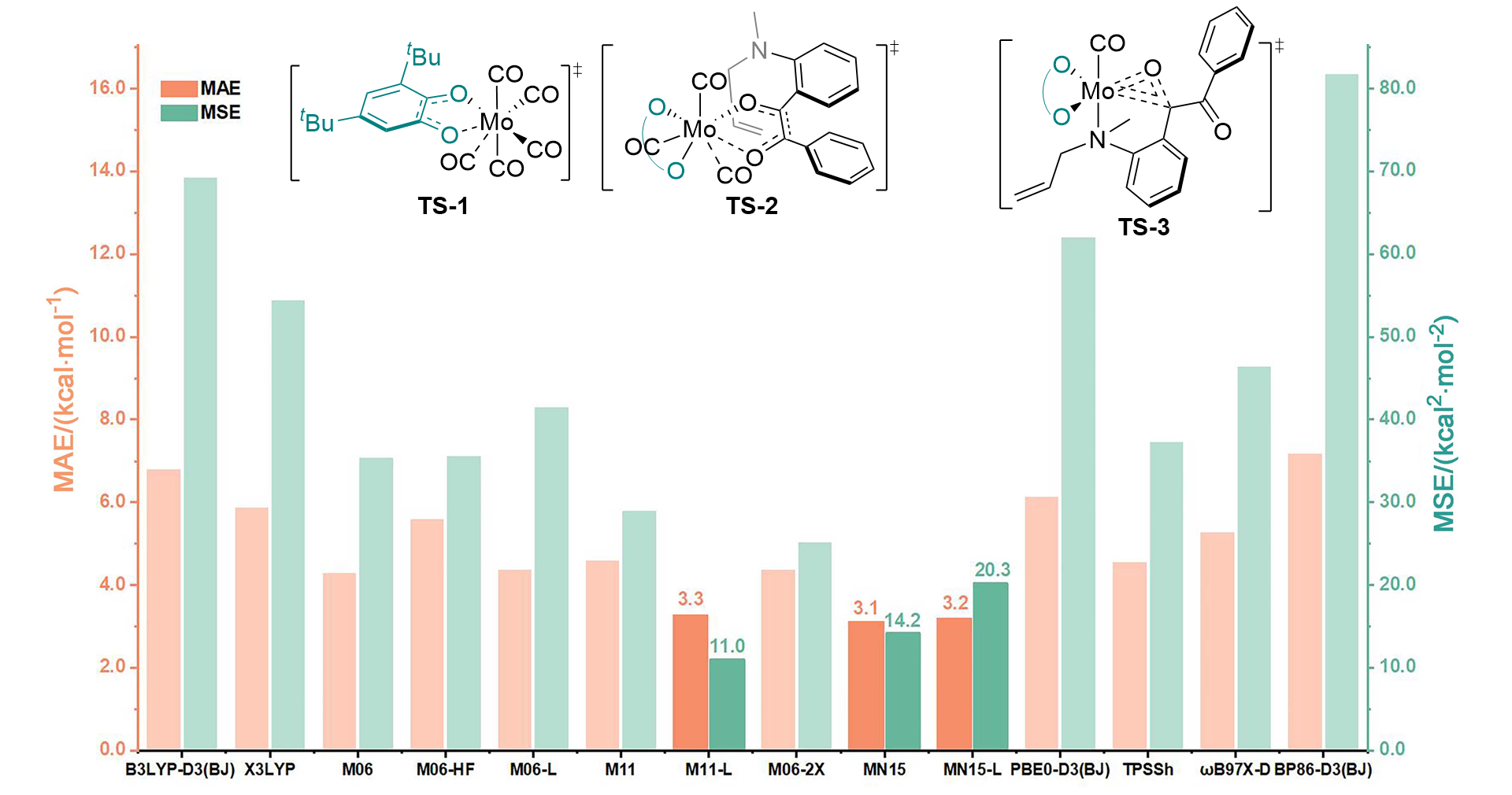
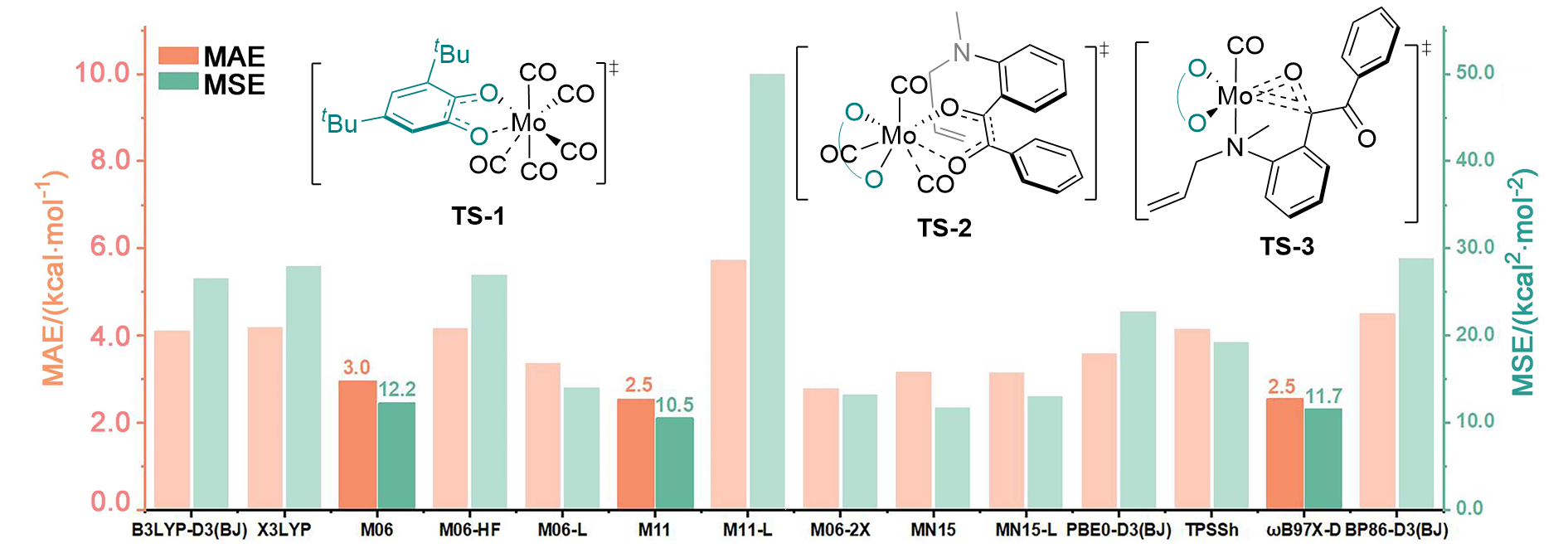
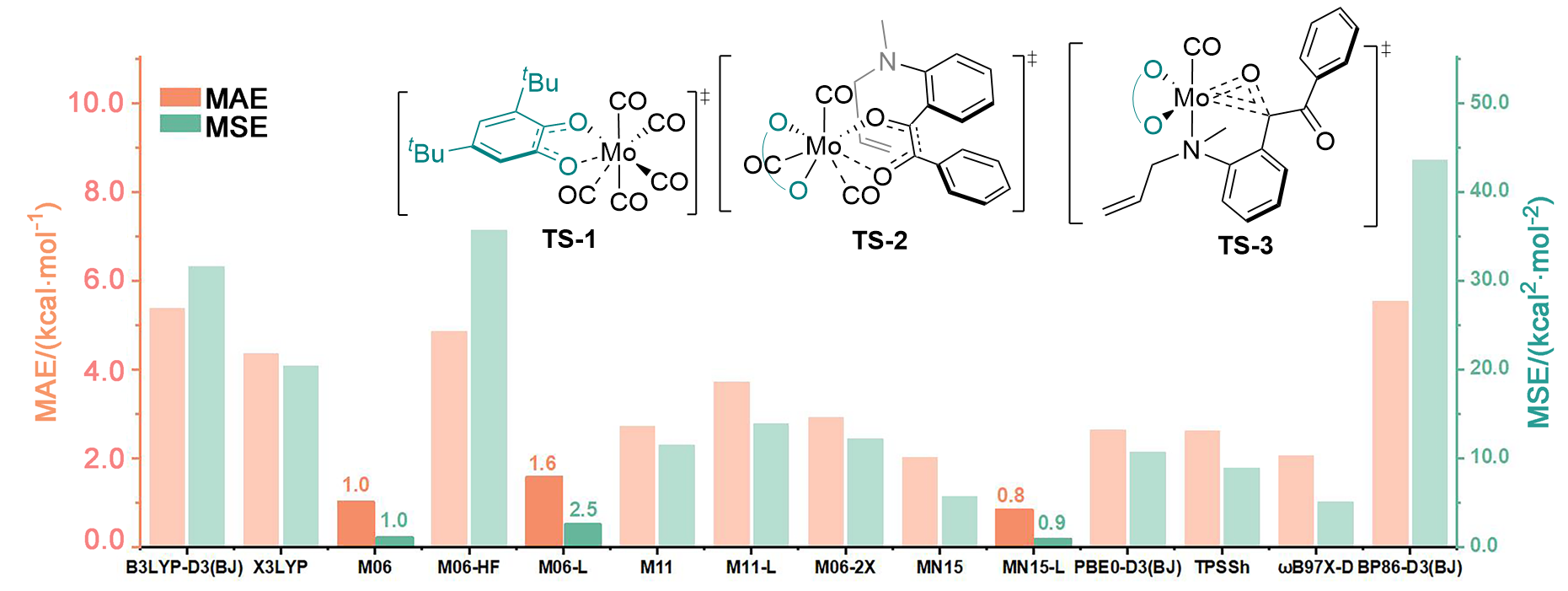
| [15] |
(a)
doi: 10.1038/s41467-019-12467-0 pmid: 31582755 |
|
(b)
doi: 10.1146/physchem.2017.68.issue-1 pmid: 31582755 |
|
| [16] |
doi: 10.1063/5.0128508 |
| [17] |
doi: 10.1103/PhysRevB.102.035129 |
| [18] |
|
| [19] |
doi: 10.1021/acs.jctc.8b00288 |
| [20] |
Frisch, M. J. T. G. W.;
|
| [21] |
(a)
doi: 10.1002/wcms.v2.1 |
|
(b)
|
|
|
(c)
doi: 10.1063/5.0004608 |
|
| [1] |
(a)
doi: 10.1093/advances/nmx001 pmid: 29767695 |
|
(b)
doi: 10.6023/cjoc201707017 pmid: 29767695 |
|
|
(胡传峰, 周建豪, 黄志达, 傅惠惠, 彭新华, 有机化学, 2018, 38, 486.)
doi: 10.6023/cjoc201707017 pmid: 29767695 |
|
|
(c)
pmid: 29767695 |
|
|
(宋礼成, 罗春成, 有机化学, 2001, 21, 1009.)
pmid: 29767695 |
|
| [2] |
doi: 10.1016/j.ccr.2011.01.027 |
| [3] |
(a)
pmid: 12114025 |
|
(b)
doi: 10.1016/s0968-0004(02)02107-2 pmid: 12114025 |
|
|
(c)
pmid: 12114025 |
|
| [4] |
(a)
doi: 10.1038/nchem.2011 pmid: 25054948 |
|
(b)
doi: 10.1002/chem.v25.1 pmid: 25054948 |
|
| [5] |
(a)
doi: 10.1002/anie.v45:23 pmid: 27126041 |
|
(b)
doi: 10.1126/science.aaf4622 pmid: 27126041 |
|
| [22] |
doi: 10.1016/j.trechm.2020.02.005 |
| [23] |
doi: 10.1103/PhysRevB.33.8822 |
| [24] |
(a)
doi: 10.1021/j100096a001 |
|
(b)
doi: 10.1063/1.464913 |
|
| [25] |
doi: 10.1063/1.478522 |
| [26] |
doi: 10.1016/j.cplett.2015.12.069 |
| [27] |
doi: 10.1063/1.3382344 |
| [28] |
doi: 10.1039/b810189b |
| [29] |
doi: 10.1063/1.2370993 |
| [30] |
doi: 10.1021/jz201525m |
| [31] |
doi: 10.1021/acs.jctc.5b01082 |
| [5] |
(c)
doi: 10.1021/jacs.3c10430 pmid: 27126041 |
| [6] |
doi: 10.1021/jacs.7b03070 pmid: 28414434 |
| [7] |
doi: 10.1002/chem.v10:2 |
| [8] |
(a)
doi: 10.1021/cr400443z pmid: 34045715 |
|
(b)
doi: 10.1038/s41557-021-00701-6 pmid: 34045715 |
|
|
(c)
doi: 10.1002/anie.v53.43 pmid: 34045715 |
|
|
(d)
doi: 10.1039/D0CS00923G pmid: 34045715 |
|
| [9] |
(a)
doi: 10.1007/s12274-022-5277-3 |
|
(b)
doi: 10.1016/j.nanoen.2019.04.060 |
|
| [32] |
doi: 10.1007/s00214-007-0310-x |
| [33] |
doi: 10.1021/jz201170d |
| [34] |
doi: 10.1039/C6SC00705H |
| [35] |
(a)
doi: 10.1103/PhysRevLett.91.146401 |
|
(b)
doi: 10.1063/1.1795692 |
|
| [36] |
doi: 10.1073/pnas.0308730100 |
| [37] |
doi: 10.1063/5.0206533 |
| [38] |
(a)
doi: 10.1039/b508541a |
|
(b)
doi: 10.1039/b515623h |
|
| [39] |
doi: 10.1021/jp810292n |
| [10] |
doi: 10.1002/anie.v60.28 |
| [11] |
doi: 10.1039/D3QO00567D |
| [12] |
doi: 10.6023/cjoc202405018 |
|
(孙庆浩, 鲍晓光, 有机化学, 2024, 44, 3518.)
doi: 10.6023/cjoc202405018 |
|
| [13] |
(a)
doi: 10.1002/advs.v11.47 |
|
(b)
doi: 10.1021/acscatal.6b02523 |
|
| [14] |
(a)
doi: 10.1007/s11051-017-4072-7 pmid: 29206033 |
|
(b)
doi: 10.1021/acs.inorgchem.7b02133 pmid: 29206033 |
|
| [40] |
(a)
doi: 10.1002/jcc.v29:2 |
|
(b)
doi: 10.1007/s00214-007-0250-5 |
|
| [41] |
|
| [42] |
doi: 10.1021/acscatal.1c02956 |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
