Chinese Journal of Organic Chemistry ›› 2021, Vol. 41 ›› Issue (7): 2666-2675.DOI: 10.6023/cjoc202012037 Previous Articles Next Articles
REVIEWS
谭胖a,b, 刘旭红a,c, 谌彤童d, 秦智慧a,b, 杨涛a, 刘晓彤a,e,f,g, 刘秀磊a,b,*( )
)
收稿日期:2020-12-22
修回日期:2021-02-08
发布日期:2021-03-04
通讯作者:
刘秀磊
基金资助:
Pang Tana,b, Xuhong Liua,c, Tongtong Chend, Zhihui Qina,b, Tao Yanga, Xiaotong Liua,e,f,g, Xiulei Liua,b()
Received:2020-12-22
Revised:2021-02-08
Published:2021-03-04
Contact:
Xiulei Liu
Supported by:Share
Pang Tan, Xuhong Liu, Tongtong Chen, Zhihui Qin, Tao Yang, Xiaotong Liu, Xiulei Liu. Research Progress on New Organic Molecules Design via Machine Learning[J]. Chinese Journal of Organic Chemistry, 2021, 41(7): 2666-2675.




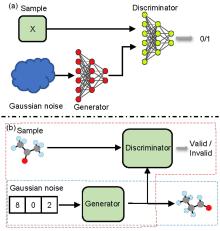




| Method | Accuracy | Accuracy without padding | LogP | QED | SAS |
|---|---|---|---|---|---|
| Without BASE64 | 98.14% | 95.08% | 0.928 | 0.093 | 0.444 |
| BASE64 | 98.53% | 96.33% | 0.934 | 0.092 | 0.440 |
| Method | Accuracy | Accuracy without padding | LogP | QED | SAS |
|---|---|---|---|---|---|
| Without BASE64 | 98.14% | 95.08% | 0.928 | 0.093 | 0.444 |
| BASE64 | 98.53% | 96.33% | 0.934 | 0.092 | 0.440 |
| [1] |
(a) Notomi, M.; Naganuma, M.; Nishida, T.; Tamamura, T.; Iwamura, H.; Nojima, S.; Okamoto, M. Appl. Phys. Lett. 1991, 58,720.
doi: 10.1063/1.104526 |
|
(b) Drews, J. Science 2000, 287(5460),1960.
doi: 10.1126/science.287.5460.1960 |
|
| [2] |
(a) Zgou, H.; Hamidi, M.; Lére-Porte,J. -P.; Serein-Spirau, F.; Bouachrine, M. Acta Phys.-Chim. Sin. 2008, 24(1),37.
doi: 10.1016/S1872-1508(08)60003-0 |
|
(b) Qian, L.; Shen, Y.; Chen, J.; Zheng, K. Acta. Phys.-Chim. Sin. 2006, 22(11),1372.
doi: 10.1016/S1872-1508(06)60069-7 |
|
| [3] |
Langer, M.; Goeßmann, A.; Rupp, M. arXiv: 2003.12081.
|
| [4] |
(a) Bohacek, R.; McMartin, C.; Guida, W. Med. Res. Rev. 1996, 16,3.
doi: 10.1002/(ISSN)1098-1128 |
|
(b) Ruddigkeit, L.; van Deursen, R.; Blum,L. C.; Reymond,J. -L. J. Chem. Inf. Model. 2012, 52(11),2864.
doi: 10.1021/ci300415d |
|
| [5] |
DiMasi,J. A.; Grabowski,H. G.; Hansen,R. W. J. Health Econ. 2016, 47,20.
doi: 10.1016/j.jhealeco.2016.01.012 |
| [6] |
(a) Tan, N.; Li, J.; Li, Z.; Li, X. Acta. Phys.-Chim. Sin. 2006, 22(4),397.
doi: 10.1016/S1872-1508(06)60011-9 |
|
(b) LeCun, Y.; Bengio, Y.; Hinton, G. Nature 2015, 521(7553),436.
doi: 10.1038/nature14539 |
|
|
(c) Eraslan, G.; Avsec, Ž.; Gagneur, J.; Theis,F. J. Nat. Rev. Genet. 2019, 20(7),389.
|
|
|
(d) Burbidge, R.; Trotter, M.; Buxton, B.; Holden, S. Comput. Chem. 2001, 26(1),5.
doi: 10.1016/S0097-8485(01)00094-8 |
|
|
(e) Liu, X.; Zhang, T.; Yang, T.; Liu, X.; Song, X.; Yang, Y.; Li, N.; Rignanese,G. -M.; Li, Y.; Wen, X. J. Phys. Chem. A 2020, 124(42),8866.
doi: 10.1021/acs.jpca.0c06319 |
|
| [7] |
(a) Williams,R. J.; Zipser, D. Neural Comput. 1989, 1(2),270.
doi: 10.1162/neco.1989.1.2.270 |
|
(b) Yu, S.; Su, J.; Luo, D. IAcc 2019, 7,176600.
|
|
| [8] |
(a) Van Houdt, G.; Mosquera, C.; Nápoles, G. Artif. Intell. Rev. 2020.
|
|
(b) Hou, H.; Xu, I.; Chen, M.; Liu, Z.; Guo, W.; Gao, M.; Xin, Y.; Cui, L. IAcc 2020, 8,90907.
|
|
| [9] |
(a) Cong, I.; Choi, S.; Lukin, M. Nat. Phys. 2019, 15,1.
|
|
(b) Soydemir, D. Int. J. Intell. Syst. Appl. Eng. 2019, 7,222.
doi: 10.18201/ijisae.2019457674 |
|
| [10] |
Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Yu, P. IEEE T. Neur. Net. Lear. 2021, 32,4.
|
| [11] |
Sanchez, B.; Aspuru-Guzik, A. Science 2018, 361,360.
doi: 10.1126/science.aat2663 |
| [12] |
Schwalbe-Koda, D.; Gómez-Bombarelli, R. arXiv: 1907.01632v1.
|
| [13] |
Herbst, I. Commun. Math. Phys. 1974, 35,181.
doi: 10.1007/BF01646192 |
| [14] |
Arora, A. Can. Med. Assoc. J. 2020, 192,E848.
doi: 10.1503/cmaj.200092 |
| [15] |
Meyer, F. Pattern Recogn. Lett. 2014, 47,72.
doi: 10.1016/j.patrec.2014.02.018 |
| [16] |
(a) Karabunarliev, S.; Ivanov, J.; Mekenyan, O. Comput. Chem. 1994, 18,189.
doi: 10.1016/0097-8485(94)85010-0 |
|
(b) Lin,T. -S.; Coley, C.; Mochigase, H.; Beech, H.; Wang, W.; Wang, Z.; Woods, E.; Craig, S.; Johnson, J.; Kalow, J.; Jensen, K.; Olsen, B. ACS Cent. Sci. 2019,5.
|
|
| [17] |
Arsham,D. H.; Davani, D.; Yu, J. Math. Comput. Simulat. 1993, 35,493.
doi: 10.1016/0378-4754(93)90067-5 |
| [18] |
O'Boyle, N. J. Cheminform. 2012, 4,22.
doi: 10.1186/1758-2946-4-22 |
| [19] |
Walsh,M. O. Am. Book Rev. 2012, 33,23.
|
| [20] |
Ikebata, H.; Hongo, K.; Isomura, T.; Maezono, R.; Yoshida, R. J. Comput.-Aided Mater. Des. 2017,31.
|
| [21] |
Ertl, P.; Lewis, R.; Martin, E.; Polyakov, V. arXiv: 1712.07449v2.
|
| [22] |
(a) Heller, S.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. J. Cheminform. 2015,7.
|
|
(b) Grethe, G.; Blanke, G.; Kraut, H.; Goodman, J. J. Cheminform. 2018,10.
|
|
| [23] |
Gómez-Bombarelli, R.; Duvenaud, D.; Hernández-Lobato, J.; Aguilera-Iparraguirre, J.; Hirzel, T.; Adams, R.; Aspuru-Guzik, A. ACS Cent. Sci. 2016,4.
|
| [24] |
(a) Bolano, A. Sci. Trends 2018.
|
|
(b) Meziani, A. Proc. Am. Math. Soc. 1996,124.
|
|
|
(c) Sandi-Urena, S. Chem. Tea. Inter. 2019.
|
|
| [25] |
Lusci, A.; Pollastri, G.; Baldi, P. J. Chem. Inf. Model. 2013,53.
|
| [26] |
Duvenaud, D.; Maclaurin, D.; Aguilera-Iparraguirre, J.; Gómez- Bombarelli, R.; Hirzel, T.; Aspuru-Guzik, A.; Adams, R. Adv. Neural Inf. Process. Syst. 2015,13.
|
| [27] |
Chen, C.; Ye, W.; Zuo, Y.; Zheng, C.; Ong,S. P. Chem. Mater. 2019, 31(9),3564.
doi: 10.1021/acs.chemmater.9b01294 |
| [28] |
Ramakrishnan, R.; Dral,P. O.; Rupp, M.; von Lilienfeld,O. A. Sci. Data 2014, 1(1),140022.
doi: 10.1038/sdata.2014.22 |
| [29] |
RDKit: http://www.rdkit.org
|
| [30] |
OpenSMILES:http://opensmiles.org/
|
| [31] |
MolVS: https://molvs.readthedocs.io/en/latest/
|
| [32] |
Turcani, L.; Berardo, E.; Jelfs, K. J. Comput. Chem. 2018, 39,1931.
doi: 10.1002/jcc.v39.23 |
| [33] |
Liu, Y.; Lin, S.; Clark, R. Proc. AAAI Con. Artif. Intel. 2020, 34,13869.
|
| [34] |
Thada, D.; Shrivastava, U.; Sharma, J.; Singh, K.; Ranadeep, M. Int. J. Inn. Res. Com. Sci. Tech. 2020,8.
|
| [35] |
(a) Hinton, G.; Zemel, R. Adv. Neural Inf. Process. Syst. 1994,6.
|
|
(b) Soydaner, D. Neural Pro. Lett. 2020.
|
|
|
(c) Ferreira, D.; Silva, S.; Abelha, A.; Machado, J. Appl. Sci. 2020, 10,5510.
doi: 10.3390/app10165510 |
|
| [36] |
Ponti, M.; Kittler, J.; Riva, M.; de Campos, T.; Zor, C. Pattern Recogn. 2017,61.
|
| [37] |
Burda, Y.; Grosse, R.; Salakhutdinov, R. arXiv: 1509.00519v4.
|
| [38] |
Kulkarni, T.; Whitney, W.; Kohli, P.; Tenenbaum, J. arXiv:1503.03167v4.
|
| [39] |
Arjovsky, M.; Chintala, S.; Bottou, L. arXiv:1701.07875v3.
|
| [40] |
Rueschendorf, L. Probab. Theory Rel. Fields 1985, 70,117.
doi: 10.1007/BF00532240 |
| [41] |
Lamberti, P.; Majtey, A. Phys. A 2003, 329,81.
|
| [42] |
Miyato, T.; Kataoka, T.; Koyama, M.; Yoshida, Y. arXiv: 1802.05957v1.
|
| [43] |
Harding, A.; Mine, H.; Osaki, S. Oper. Res. Q. (1970-1977) 1971, 22,192.
|
| [44] |
Lek, S.; Park,Y. S. Encycl. Ecol. 2008,2455.
|
| [45] |
(a) Guimaraes, G.; Sanchez, B.; Farias, P.; Aspuru-Guzik, A. 2017.
|
|
(b) Popova, M.; Isayev, O.; Tropsha, A. Sci. Adv. 2017,4.
|
|
| [46] |
Kusner, M.; Paige, B.; Hernández-Lobato, J. arXiv: 1703.01925v1.
|
| [47] |
(a) Johansen, S.; Juselius, K. Oxford Bull. Econ. Statist. 1990, 52,169.
doi: 10.1111/obes.1990.52.issue-2 |
|
(b) Chow, G. Econ. Modelling 1984, 1,134.
doi: 10.1016/0264-9993(84)90001-4 |
|
| [48] |
Dai, H.; Tian, Y.; Dai, B.; Skiena, S.; Song, L. arXiv: 1802.08786v1.
|
| [49] |
De Cao, N.; Kipf, T. aarXiv: 1805.11973v1.
|
| [50] |
Jin, W.; Barzilay, R.; Jaakkola, T. arXiv: 1802.04364.
|
| [51] |
Jørgensen, M.; Mortensen, H.; Meldgaard, S.; Kolsbjerg, E.; Jacobsen, T.; Sørensen, K.; Hammer, B. J. Chem. Phys. 2019, 151,054111.
doi: 10.1063/1.5108871 |
| [52] |
Noh, J.; Kim, J.; Stein,H. S.; Sanchez-Lengeling, B.; Gregoire,J. M.; Aspuru-Guzik, A.; Jung, Y. Matter 2019, 1(5),1370.
doi: 10.1016/j.matt.2019.08.017 |
| [53] |
Jain, A.; Ong,S. P.; Hautier, G.; Chen, W.; Richards,W. D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; Persson,K. A. APL Mater. 2013, 1(1),011002.
doi: 10.1063/1.4812323 |
| [54] |
Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez,A. N.; Kaiser, Ł.; Polosukhin, I. In Proceedings of the 31st International Conference on Neural Information Processing Systems, Curran Associates Inc., Long Beach, California, USA, 2017, p. 6000.
|
| [1] | Liu Yidi, Yang Qi, Li Yao, Zhang Long, Luo Sanzhong. Application of Machine Learning in Organic Chemistry [J]. Chinese Journal of Organic Chemistry, 2020, 40(11): 3812-3827. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
