


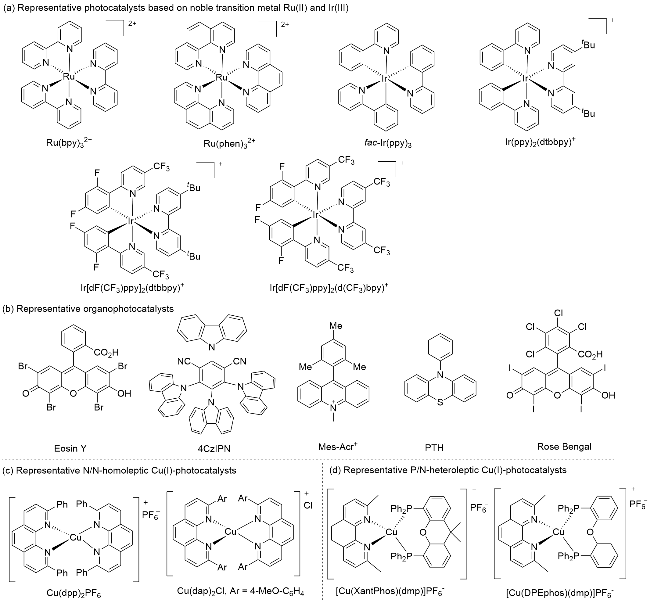
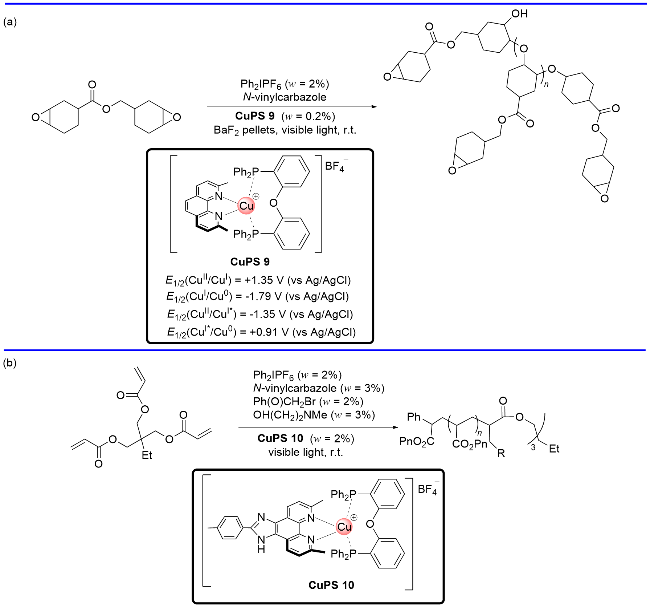
自2008年以来, 可见光驱动的光催化反应在有机化学和催化化学中获得了迅猛发展[1]. 在光催化反应中, 光催化剂扮演着非常重要的角色. 迄今为止, 占主流的过渡金属光催化剂大都集中在以钌(II)和铱(III)为代表的贵金属配合物上(图1, a). 虽然这些贵金属光催化剂有很多的优势, 如在可见光波段有很强的吸收、较长的激发态寿命及合适且可调节的氧化还原电位等, 但也存在中心金属储量少、价格昂贵及毒性高等不足, 从而限制了这些光催化剂的规模化应用. 为弥补以上不足, 同时拓展光催化剂的范围, 化学家试图设计与合成价廉及低毒的有机光催化剂来替代贵金属光催化剂(图1, b)[2], 但有机光催化剂普遍存在激发态寿命较短及光学稳定性差等缺陷. 另一方面, 化学家也试图开发基于丰产金属(如铜、铬、铁、钴等)为中心金属的光催化剂[3]. 其中, 鉴于金属铜具有储量丰富、价格低、毒性小及催化活性高等优点, 铜(I)基光催化剂的开发和应用受到特别的关注[4]. 在Cu(I)光催化剂的发展过程中, 早期的Cu(I)基光催化剂主要是一些N/N均配的铜(I)配合物(图1, c). 近年来, P/N杂配铜(I)光催化剂的开发和应用异军突起(图1, d, Scheme 1), 在可见光驱动的光催化反应中发挥了重要作用, 取得了很大进展.
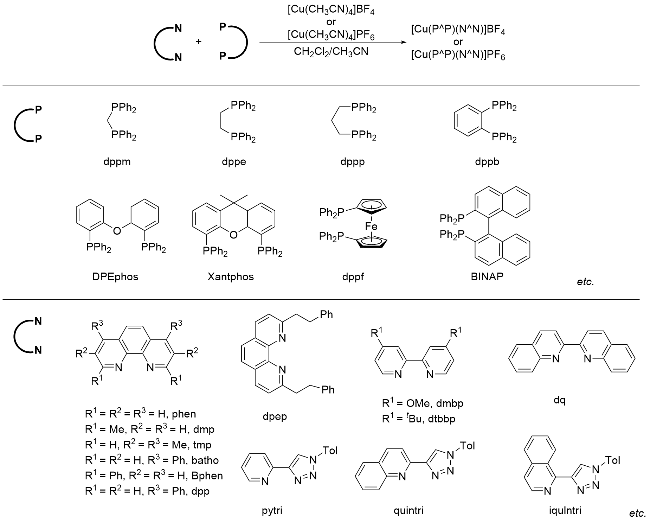
实际上, P/N杂配铜(I)光催化剂的研究最早可追溯到20世纪80年代末. McMillin课题组[5]率先合成了一系列单齿膦配体和菲啰啉杂配的铜(I)络合物, 但这类配合物的光激发态性能很差. 直到2002年, Walton和McMillin课题组[6]发现由带有一定位阻的双齿膦配体与菲啰啉配体螯合生成的Cu(I)光催化剂, 其激发态寿命和荧光量子产率有显著提升, 性能甚至大大优于N/N均配的铜(I)光催化剂, 如双菲啰啉均配的Cu(dap)2Cl. 随后, Collins[7]、Beller[8]、支志明[9]和Evano[10]等学者的开创性研究证明了P/N杂配Cu(I)光催化剂在光催化有机合成方面有巨大潜力. 基于不同电子和立体位阻效应的双膦配体和双氮配体, 可以设计与合成具有相当宽泛激发态或基态电位范围的P/N杂配铜(I)光催化剂库(Scheme 2), 从而为光催化反应条件的优化提供了极大的便利. 研究表明, 双氮配体α-位带有取代基有利于提高P/N杂配铜基光敏剂的激发态寿命[4]. 双磷配体和双氮配体带有的给电子基团有利于增加金属铜中心的电子密度, 进而增加铜催化剂的还原能力; 反之, 双磷配体和双氮配体带有的吸电子基团会降低金属铜中心的电子密度, 进而导致铜催化剂氧化能力提高[4]. 关于P/N杂配铜光催化剂的相关物理参数(如最大吸收波长、激发态寿命和能量等)和基态、激发态的氧化还原电位, Evano等[4d]在最近的一篇综述中做了非常详细的总结. 近年来的研究表明, 这类光催化剂不仅可以替代钌(II)、铱(III)等贵重金属光催化剂催化常规的光催化反应, 而且还能实现钌(II)、铱(III)等光催化剂所不能完成的光催化反应, 对于高效构建有机分子多样性发挥了不可替代的作用(Scheme 3). 本文根据反应机理的不同, 从单电子转移、铜基光催化剂的双功能催化、双金属协同催化及能量转移四个类型出发, 对P/N杂配Cu(I)光催化剂介导的光催化反应进展进行综述.
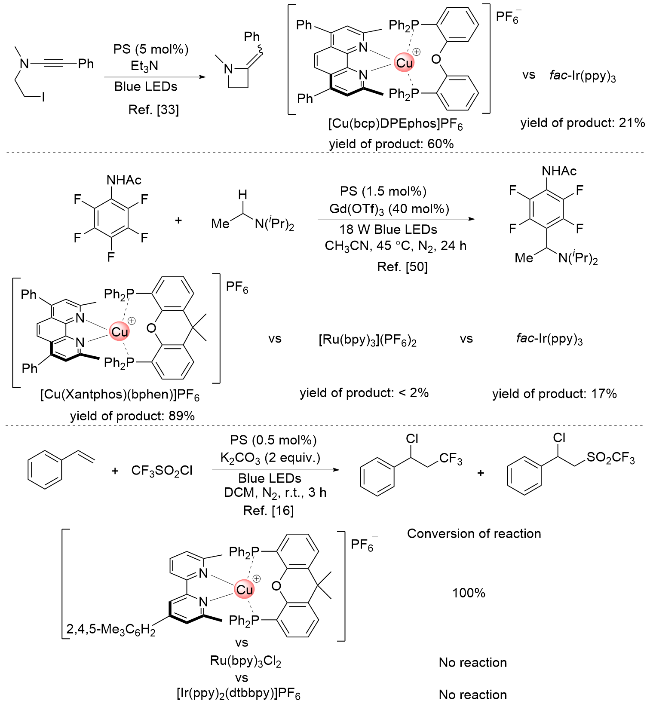
图式2 P/N杂配铜(I)光催化剂和传统贵金属光催化剂在若干光催化反应中的表现对比Scheme 2 Performance of P/N-heteroleptic Cu(I)-photocatalysts versus traditional noble-metal based photocatalysts in several photocatalytic reactions |
1 基于单电子转移的反应
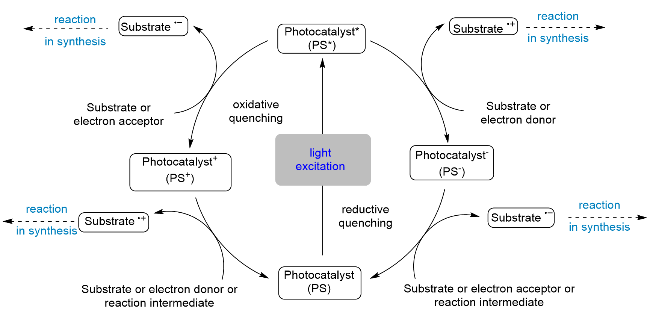
光催化反应中最为普遍的反应类型是单电子转移(SET)反应, P/N杂配铜基光催化剂介导的光催化反应也不例外. 根据光催化剂激发态与底物之间传递电子的方向不同, 单电子转移反应可以分为两种机理模式, 即氧化淬灭循环机理模式和还原淬灭循环机理模式(Scheme 3). 在氧化淬灭循环机理模式中, 激发态的光催化剂(PS*)作为还原剂首先与氧化性的反应底物发生单电子转移启动光反应, 自身转化为氧化态的光催化剂(PS+). 然后, PS+再与反应体系中的还原性底物或中间体发生单电子转移反应, 再生基态光催化剂, 完成催化循环; 而在还原淬灭循环机理模式中, 激发态的光催化剂(PS*)则作为氧化剂首先与还原性的反应底物发生单电子转移启动光反应, 自身转化为还原态的光催化剂 (PS-), 然后被反应体系中的氧化性底物或中间体单电子氧化, 再生基态催化剂并完成催化循环. 得益于P/N杂配铜(I)光催化剂的氧化还原电位易于通过双磷和双氮配体的电子和立体位阻效应进行调控的优势, 迄今为止, 化学家们已经设计和合成了庞大的包含各种氧化还原电位的铜(I)光催化剂库, 从而为不同机理模式进行的单电子转移反应提供了多样性的选择.
2019年, Poisson课题组[11]报道了在光照射下以及[Cu(DPEphos)(DMEGqu)]PF6存在下, 通过单电子转移途径, 将芳基卤(溴、碘)化物与B2Pin2转化为芳基硼试剂的反应(Scheme 4, a). 2020年, Fimognari课题组[12]以[Cu(BINAP)(dq)]BF4 (CuPS 2)为光催化剂, 实现了N-杂环磺酰胺类化合物的脱磺酰基反应(Scheme 4, b). 有趣的是, 这两种反应无论从氧化淬灭循环还是从还原淬灭循环机理均能实现. 然而, 大多数P/N杂配Cu(I)光催化剂介导的单电子转移反应只能经历其中一种催化循环模式. 下面将分别从氧化淬灭和还原淬灭循环模式出发, 对相关研究进行归类综述.
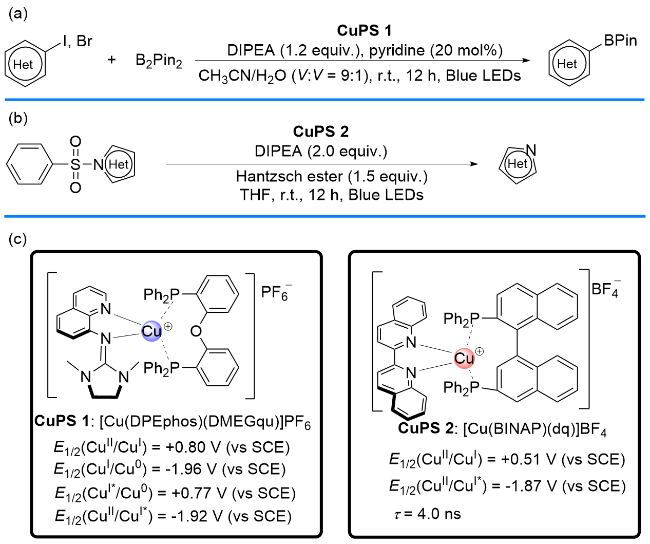
1.1 基于氧化淬灭循环机理的单电子转移反应
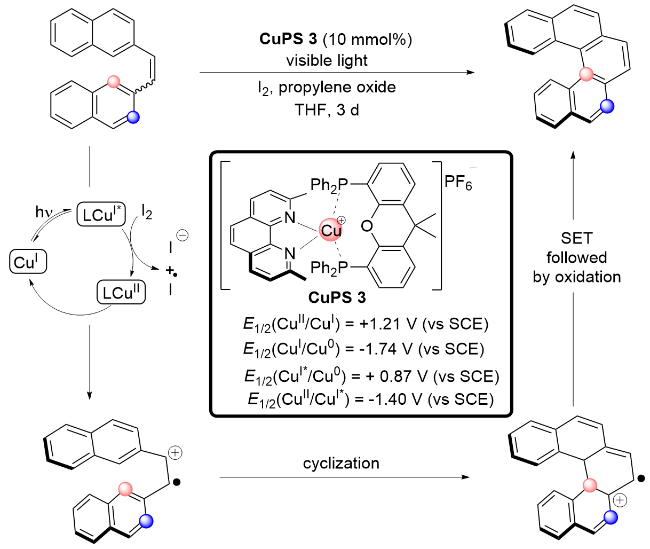
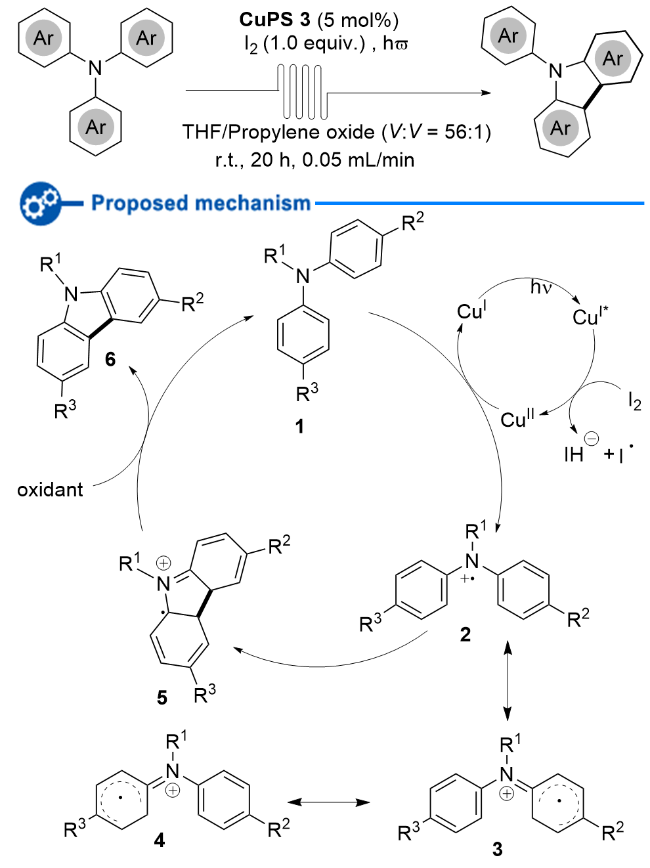
2013年, Collins课题组[13]又发展了一种以[Cu- (Xantphos)(dmp)]PF6为光敏剂, 通过连续流的方式, 有效地催化三芳基胺发生分子内氧化环化, 合成咔唑类化合物的方法(Scheme 6). 该方法通过使用连续流动条件大大加快了反应速率. 遗憾的是, 该方法仅适用于富电子的三芳基胺和N-烷基取代的二芳基胺; 当使用非对称的三芳基胺作为反应底物时, 只能得到多种异构体的混合物. 该反应可能的反应机理如下: 激发态*Cu(I)与I2相互作用产生Cu(II)和碘自由基, 接着Cu(II)通过单电子转移从三芳基叔胺1上攫取一个电子, 从而产生三芳基胺自由基正离子2; 自由基正离子2的自由基可能离域至芳环, 形成自由基正离子3或4; 随后发生分子内自由基环化得到中间体5, 5被碘自由基或氧气氧化即可得到目标产物6.
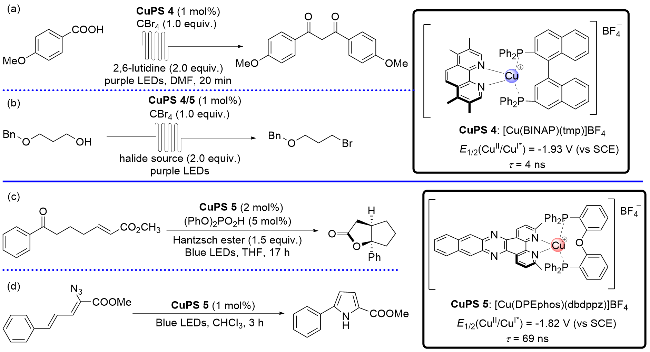
随后, Collins课题组又相继设计与合成了两类不同类型的P/N杂配络合物, 并以其为光敏剂开发了几个新反应. 2018年, 该课题组[14]在紫光照射以及[Cu(BINAP)- (tmp)]BF4存在下, 通过连续流的反应方式, 实现了由芳基羧酸合成1,3-二羰基化合物(Scheme 7, a)及由烷基醇合成烷基溴代物(Scheme 7, b). 接着, 该课题组[15]通过调控双氮配体结构进而开发出新的铜基光催化剂([Cu-(DPEphos)(dbdppz)]BF4). 在该光催化剂的作用下, 通过光氧化还原成功地构建了环戊烷[b]并呋喃-2-酮类化合物(Scheme 7, c)以及5-苯基-1H-吡咯-2-羧酸甲酯类化合物(Scheme 7, d).
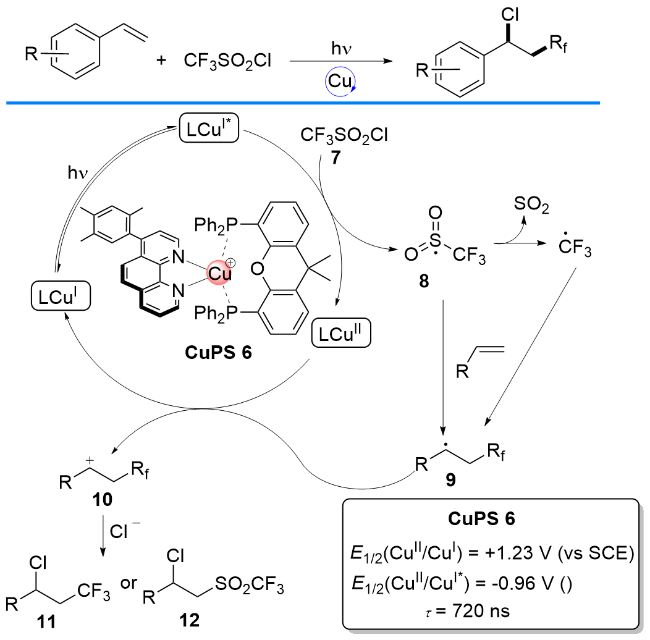
2018年, 胡喜乐课题组[16]开发出一种4,6-二取代- 2,2'-联吡啶与Xantphos配位的铜基络合物(Scheme 8), 与广泛使用的N/N均配铜(I)催化剂[Cu(dap)2]Cl[17-19]相比, 该络合物表现出更长的激发态寿命和更高的Cu(I)/ Cu(II)电势. 其催化苯乙烯的氯化三氟甲基化反应尤为有效, 实现了光催化条件下苯乙烯的氯化三氟甲基化反应. 作者提出了可能的反应机理: 三氟甲烷磺酰氯7通过单电子转移过程从P/N杂配Cu(I)光敏剂上攫取一个电子, 从而得到自由基8及自由基8脱去SO2后的三氟甲基自由基; 随后, 两者与末端烯烃发生自由基加成生成相应的中间体9, 然后自由基9被Cu(II)单电子氧化得到正离子物种10; 最后, 正离子10与反应体系中的游离氯负离子结合, 最终形成双官能化产物11或12.
2018年, 许兆青课题组[20]报道了P/N杂配Cu(I)络合物为光催化剂催化的甘氨酸及多肽中甘氨酸残基 Cα—H键的烷基化反应, 合成了非天然α-氨基酸. 该反应具有条件温和、官能团普适性好以及产率良好等优点. 此外, 通过简单的操作, α-烷基化的非天然氨基酸可以直接用于制备基于多肽的生物活性分子, 例如胶原三肽的类似物.
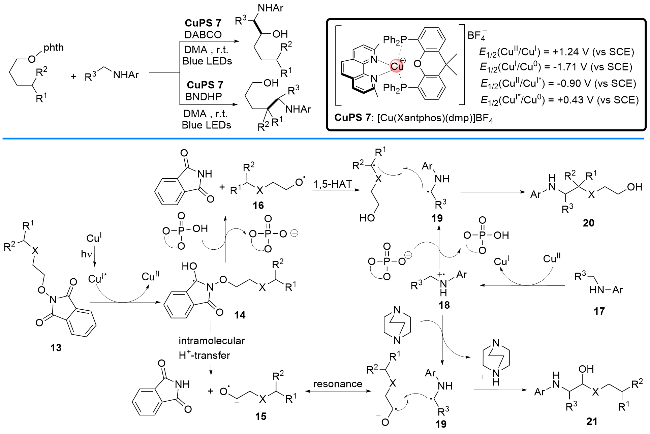
2019年, 段伟良课题组[21]报道了在铜基光催化剂[Cu(Xantphos)(dmp)]BF4存在和光照射下, 通过光氧化还原过程实现N-烷氧基邻苯二甲酰亚胺与氨基酸酯或氨基酸的交叉偶联反应(Scheme 9). 该反应通过加入不同的添加剂, 如联萘酚磷酸酯(BNDHP)或者三乙烯二胺(DABCO), 从而可以实现反应途径的调控, 选择性地得到不同的产物. 作者提出了可能的反应机理, N-烷氧基邻苯二甲酰亚胺13与P/N杂配Cu(I)光催化剂发生单电子转移反应生成14. 随后, 反应因外部添加剂的不同而经历两种不同的路径, 最后得到两种不同的产物. 其一, 在BNDHP的参与下主要生成氧基自由基物种16, 经分子内1,5-氢迁移产生碳基自由基, 并与反应体系产生的α-胺基自由基19偶联, 最后生成伯醇类产物20. 其二, 在DABCO参与下主要生成自由基负离子物种15, 然后与反应体系中形成的α-胺基自由基19偶联, 形成最终产物21.
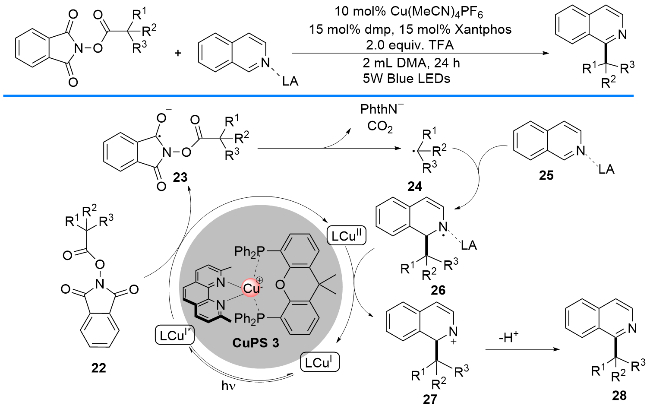
2019年, 汪清民课题组[22]报道了以P/N杂配Cu(I)络合物为光催化剂, 通过可见光诱导催化N-(酰氧基)邻苯二甲酰亚胺酯(NHPI酯)经裂解脱羧产生烷基自由基, 并与N-杂芳烃进行偶联的Minsci反应(Scheme 10). 该反应能够使各种NHPI酯脱羧, 并与异喹啉、喹啉、吡啶、嘧啶、喹唑啉、酞嗪、菲啶和哒嗪进行偶联. 作者提出了可能的反应机理: NHPI酯22通过单电子转移过程从P/N杂配Cu(I)光催化剂上攫取一个电子, 从而转化为自由基负离子23, 接着发生β-裂解和脱羧过程, 生成烷基自由基24; 随后, 烷基自由基24与氮杂芳烃25发生典型的Minsci自由基偶联反应, 经一系列转化最终生成Nα-烷基取代的氮杂环芳烃28.
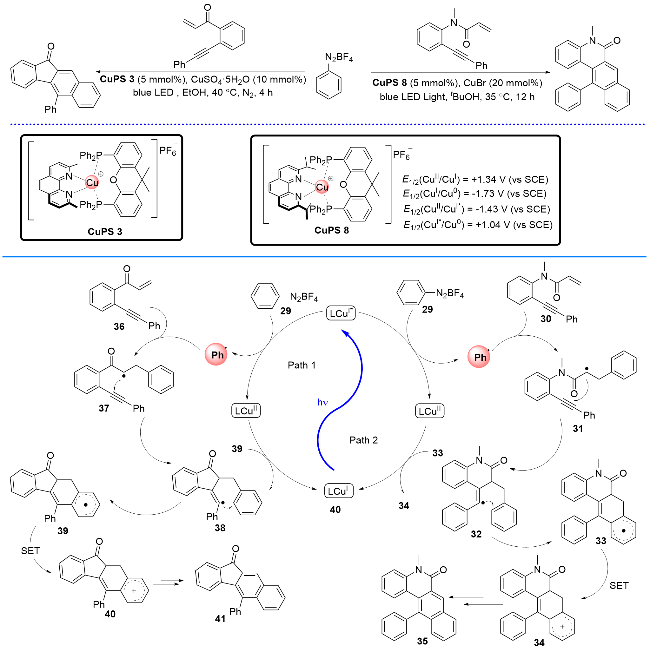
2023年, 刘运奎课题组[23-24]开发了一种以P/N杂配Cu(I)络合物为光敏剂, 以芳基重氮盐为自由基前体, 以1,n-烯炔(n=6或7)为自由基受体, 在光诱导下经多米诺串联反应来构建稠环芳烃的方法(Scheme 11). 该方法具有反应条件温和、选择性高及底物普适性好等优点. 机理研究表明, 反应首先由激发态Cu(I)*与苯基重氮盐经氧化淬灭机理启动反应, 产生苯基自由基和Cu(II)物种. 然后发生苯基自由基对1,n-烯炔的自由基加成和自由基接力环化, 形成自由基中间体33 (n=7)或者39 (n=6), 最后33或者39经单电子氧化/脱氢芳构化得到目标产物. 在该反应中, 净的结果是芳基自由基既充当了自由基供给体, 又充当了自由基接受体.
1.2 基于还原淬灭循环机理的单电子转移反应
P/N-杂配铜(I)光催化剂除了能够广泛地参与氧化淬灭循环机理启动的单电子转移反应之外, 同样在还原淬灭机理启动的自由基反应方面也取得了很大进展.
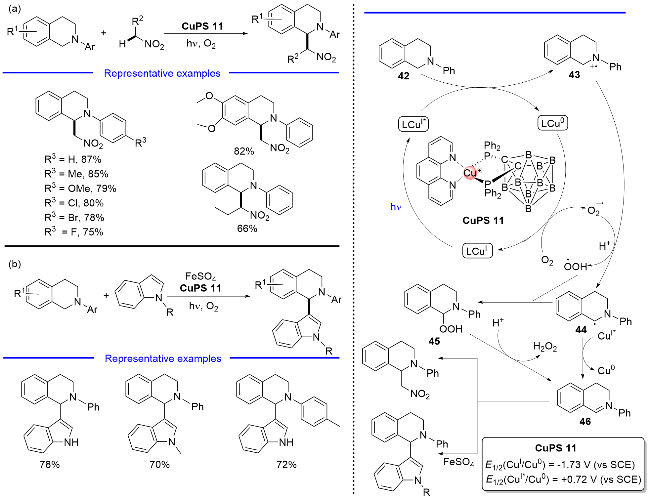
2015年, 支志明课题组[9]设计了一类含有菲啰啉和单阴离子邻碳硼烷二膦配体的P/N杂配Cu(I)络合物. 这些铜(I)络合物在可见光范围内表现出强烈的吸收, 激发态寿命达到微秒级. 其中, 菲啰啉配体的2,9-位带有甲基的Cu(I)络合物能够在可见光照条件下有效地催化四氢异喹啉与硝基烷烃之间的交叉脱氢偶联反应 (Scheme 13, a). 在硫酸亚铁的参与下该光催化剂同样能够实现四氢异喹啉与吲哚之间的氧化脱氢偶联反应, 在该反应中氧气充当了实际的氧化剂(Scheme 13, b). 作者提出了可能的反应机理: 激发态的Cu(I)*通过单电子转移过程攫取四氢异喹啉42的一个电子, 从而生成四氢异喹啉自由基阳离子43, 启动还原淬灭循环; 然后, 氧气接收Cu(0)给予的电子转化为超氧自由基负离子, 接着其与自由基阳离子43反应得到中间体45; 45在质子的辅助下脱除一分子过氧化氢转化为正离子物种46, 随后分别与硝基烷烃或吲哚进行偶联. 另外, 正离子物种46也可能直接由α-胺基自由基42与激发态的Cu(I)*经单电子氧化而得到.
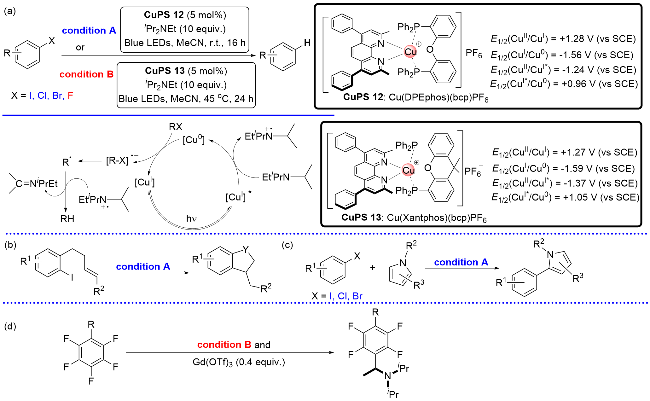
2017年, Evano课题组[10]以Cu(DPEphos)(bcp)PF6为光催化剂, 成功地实现了芳基碘或芳基溴的还原脱卤反应(Scheme 14, a), 并首次明确地提出了还原淬灭循环的机理. 隔年, Bissember课题组[27]以Cu(Xantphos)- (bcp)PF6为光催化剂, 在45 ℃下成功地实现了芳基氟的还原脱氟, 填补了芳基氟还原脱氟反应的空白(Scheme 14, a). 这两例反应均涉及还原淬灭机理启动反应, 即首先由激发态的Cu(I)*与二异丙基乙基胺发生单电子转移生成还原性的Cu(0)和胺基自由基正离子物种, 随后强还原性的Cu(0)将芳基碘(溴、氟)还原为芳基自由基, 芳基自由基通过氢原子转移(HAT)从胺基自由基正离子得到一个氢原子, 从而生成目标产物. 通过芳基自由基关键中间体, 既可以经过分子内自由基环化反应简洁地合成吲哚啉(Y=N)、二氢苯并呋喃(Y=O)以及二氢茚衍生物(Y=CH2) (Scheme 14, b), 还能以吡咯类化合物作为芳基自由基的接受体, 从而构建2-芳基取代的吡咯类衍生物(Scheme 14, c). 此外, 芳基自由基在添加剂Gd(OTf)3催化下, 能与α-胺基自由基偶联生成芳基叔胺类化合物(Scheme 14, d).
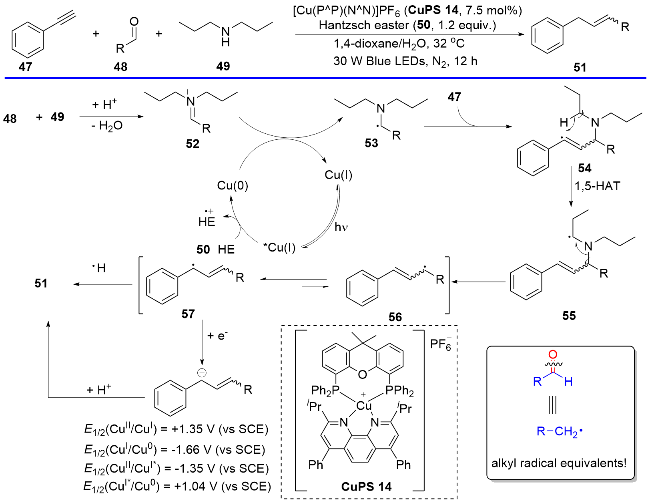
自2020以来, 刘运奎课题组[28-30]相继开发了一系列基于P/N杂配铜(I)光催化剂催化的涉及还原淬灭机理启动的光催化反应. 在这些反应中, P/N杂配铜(I)光催化剂表现出了独特的反应性能, 而传统的贵重金属光催化剂在这些反应中并没有展示很好的催化活性. 例如, 2020年, 他们[28]以P/N杂配Cu(I)光敏剂为光催化剂, 炔烃、胺以及烷基醛为底物, 实现了光催化的炔和醛脱氧偶联反应(Scheme 15). 在该反应中, 醛类化合物在胺类化合物的辅助下成为一种还原脱氧型烷基自由基等当体. 通过一系列的机理实验提出了可靠的机理过程. 首先, 在光照条件下铜光催化剂吸收光能形成激发态Cu(I)*, 紧接着Cu(I)*与汉斯酯发生单电子转移, 生成Cu(0)和汉斯酯自由基正离子, 随后, 汉斯酯自由基正离子释放质子, 促使醛和胺发生脱水缩合, 生成亚胺正离子52, 亚胺正离子被Cu(0)还原生成α-胺基自由基53, 随后与炔烃发生自由基加成得到烯基自由基中间体54, 接着发生分子内1,5-氢迁移得到自由基中间体55, 中间体55中C—N键发生均裂, 生成烯丙基自由基56, 异构化后生成更为稳定的烯丙位苄基自由基57. 最后出现两种可能途径: 第一种苄基自由基直接与汉斯酯发生氢迁移得到目标产物, 另一种则是先被Cu(0)还原成苄基负离子, 然后从体系中得到一个氢质子生成目标产物. 该反应体系温和, 采用廉价的铜配合物作为光催化剂, 炔和醛皆是廉价易得的原料, 同时底物的普适性也很高, 是一种很好的由醛构建烯烃的途径.
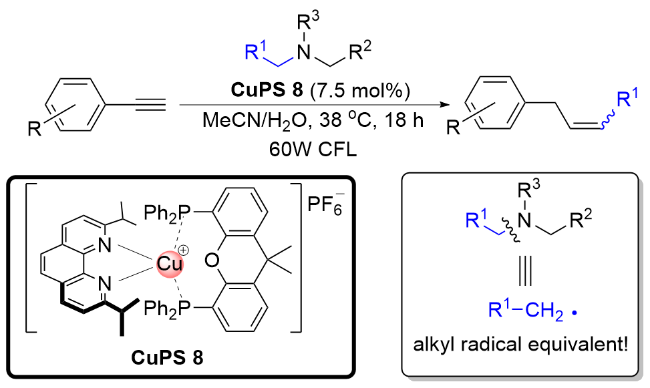
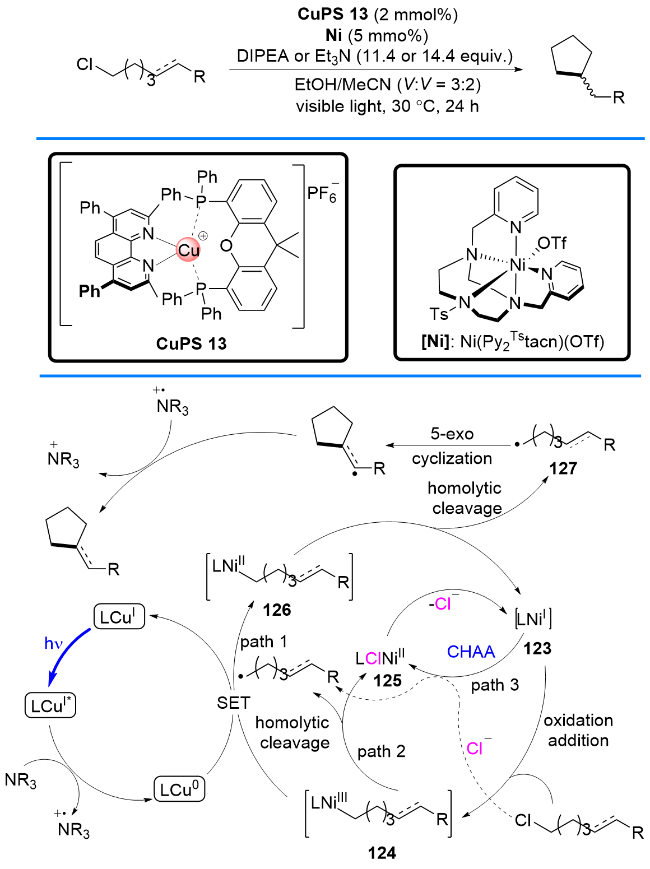
在利用P/N杂配铜基光催化剂成功实现烷基醛充当脱氧型烷基自由基等当体和叔胺充当脱胺型烷基自由基等当体的基础上, 最近, 刘运奎课题组[30]又利用P/N杂配铜基光催化剂实现了更具挑战性的课题, 即实现端烯衍生物通过碳-碳键切断充当烷基自由基等当体, 当分子内具有接受自由基的官能团存在时, 可以方便地构建环化产物(Scheme 17). 这一反应成功的关键在于利用原位产生的α-胺基自由基与烯烃发生一种全新的反应模式, 即α-胺基自由基与烯烃的复分解(交换)反应, 从而使得烯烃发生碳-碳切断成为新的自由基, 而α-胺基自由基则接受烯烃断裂的片段成为烯胺类物种. 根据作者提出的全新反应模式, 他们实现了一例1,7-烯炔的脱亚甲基化自由基环化反应. 比较有趣的是, 通过微调菲啰啉配体的结构可以改变铜基光催化剂的氧化还原电位, 从而得到不同的环化产物. 当使用还原能力较弱的铜基光催化剂CuPS 15 [E1/2(CuI/Cu0)=-1.48 V (vs SCE)]时, 得到喹啉-2-酮类化合物; 而当使用还原电位能力较强的CuPS 14 [E1/2(CuI/Cu0)=-1.66 V (vs SCE)]时, 选择性地得到3,4-二氢-喹啉-2-酮类化合物.
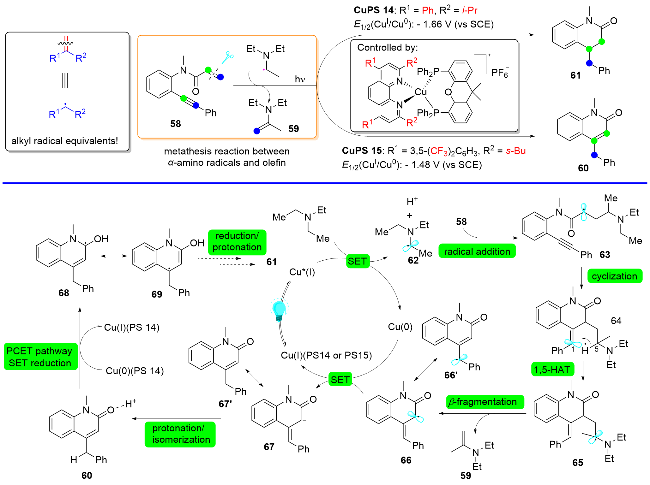
该反应的可能机理如下: 首先激发态的Cu(I)*与三正丙胺作用产生α-胺基自由基62, 62加成到1,7-烯炔58的端烯部位并引发分子内自由基接力环化形成自由基中间体64, 64发生分子内1,5-氢迁移生成一个新的α-胺基自由基65, 65易于发生自由基诱导的β-裂解, 从而引发1,7-烯炔端烯原有碳-碳键断裂形成自由基中间体66和烯胺59, 自此为止, 净的表观结果即是烯烃和α-胺基自由基62完成了复分解(交换)反应. 在还原性较弱的CuPS 15催化下, 反应选择性地得到喹啉-2-酮类化合物; 而当在还原性较强的CuPS 14催化条件下, 喹啉- 2-酮类化合物60通过质子偶合电子转移(PCET)途径, 进一步还原为3,4-二氢-喹啉-2-酮类化合物61.
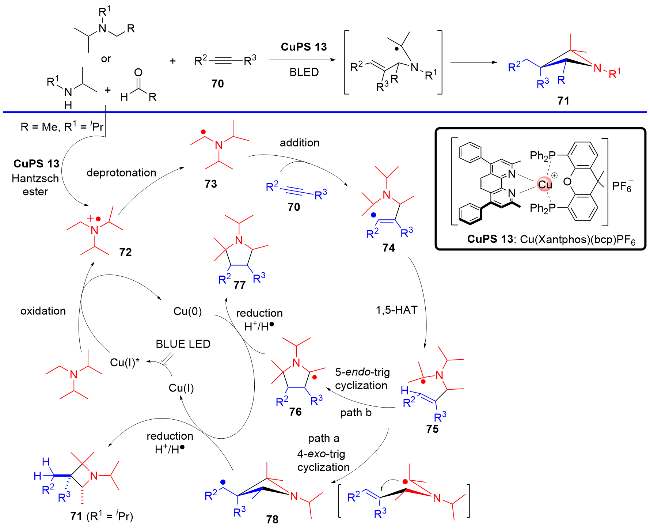
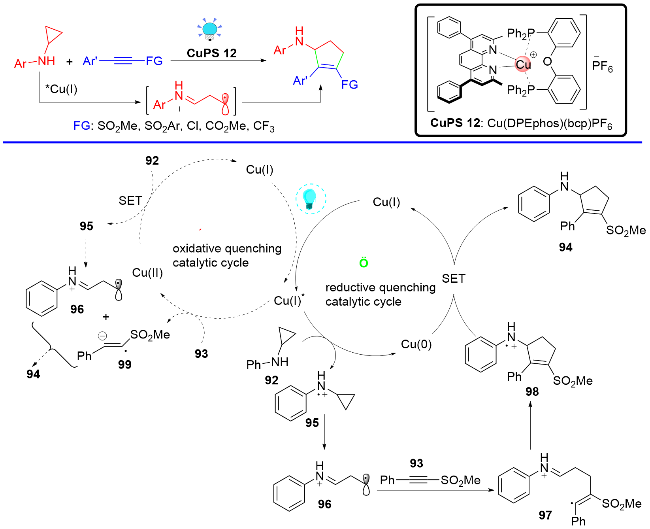
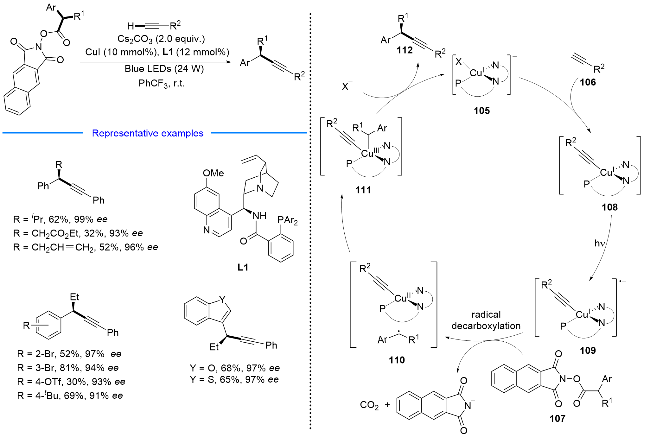
2022年, 张国柱课题组联合刘运奎课题组[31]实现了可见光诱导下P/N杂配铜(I)光催化剂催化的简单烷基胺和炔烃的[3+1]自由基串联环化反应, 简单、高效、模块化地合成了一系列氮杂环丁烷骨架(Scheme 18). 该反应也是经历现场产生的α-胺基自由基对炔烃的加成启动反应, 但与之前刘运奎课题组[28]报道的反应有所不同的是, 在该反应中采用了位阻效应较大的α-胺基自由基, 在α-胺基自由基与炔烃发生自由基加成及1,5-氢迁移之后, 并没有发生 C—N键的切断反应, 而是发生了分子内的自由基加成, 净的表观结果即是实现了烷基胺和炔烃的[3+1]自由基串联环化反应. 该方法将与药物相关的氮杂环丁烷骨架引入复杂的分子环境中, 从而为药物和天然产物衍生物的结构后修饰提供了潜在的有效手段.
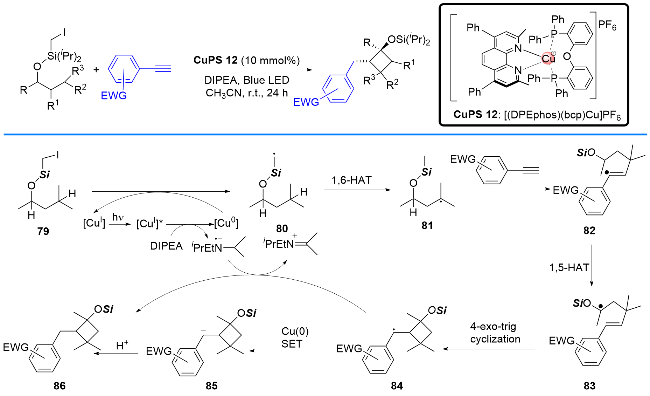
早在2021年, 张国柱课题组[32]还报道了另外一例P/N杂配Cu(I)光催化剂催化的[3+1]自由基环化反应, 成功地将碘甲基硅醚衍生物与缺电子的末端炔烃串联合成了环丁醇硅醚衍生物(Scheme 19). 作者提出了可能的反应机理: 激发态Cu(I)*首先与二异丙基乙胺发生还原淬灭生成还原态Cu(0)物种, 随后Cu(0)单电子还原碘甲基硅醚衍生物79生成碳自由基80; 接着, 该中间体发生1,6-HAT得到新的自由基81, 并与缺电子的末端炔烃加合形成自由基中间体82; 自由基82发生1,5-HAT和4-exo-trig环化过程产生自由基84. 最后, 自由基84既可以与二异丙基乙基胺自由基正离子(DIPEA•+)发生氢转移生成最终产物86, 也可以先与Cu(0)进行单电子还原生成负离子中间体64, 并经历质子化得到最终产物86.
无独有偶, Evano课题组[33]亦报道了一种以[Cu(DPEphos)(bcp)]PF6为光催化剂, 通过光氧化还原催化炔胺碘化物生成烷基自由基中间体, 再经历自由基对炔基的4-exo-trig环化, 从而合成氮杂环丁烷的反应. 同样的, 该反应首先通过P/N杂配铜光催化剂与叔胺发生还原淬灭生成还原性Cu(0)物种, 再诱导碘化物产生碳自由基中间体.
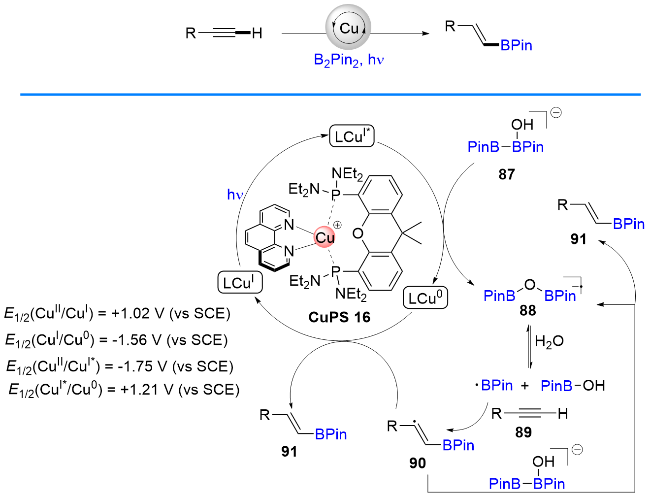
2021年, Poisson课题组[34]报道了一例可见光驱动的、P/N杂配铜基光催化剂催化的炔烃和二硼试剂B2pin2之间的自由基硼氢化反应, 生成反马加成的硼氢化产物(Scheme 20). 作者提出了可能的反应机理: 在路易斯碱的作用下, B2pin2原位转化为硼酸酯中间体[B2Pin2]OH- 87, 然后激发态的*Cu(I)被87还原淬灭生成自由基负离子物种88; 随后被水裂解为硼自由基和羟基硼试剂; 最后硼自由基对炔烃完成反马加成、自由基还原和质子化, 得到最终产物. 同年, 该课题组[35]又实现了以[Cu(XantphosTEPD)(dmp)]PF6为光催化剂催化的硅烷硼酸酯与炔烃的自由基氢硅化反应, 生成反马加成的氢硅化产物. 该反应具有反应条件温和、产率优良和底物普适性好等优点.
2 基于铜基光催化剂双功能催化的反应
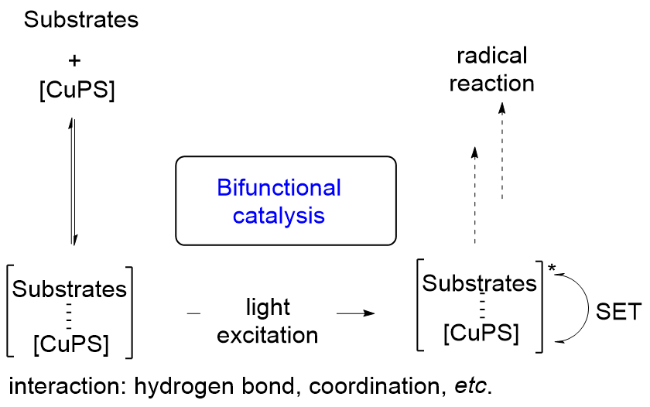
前面介绍的铜基光催化剂催化的单电子转移反应, 在反应过程中铜基光敏剂和底物之间除发生电子的转移之外, 并没有发生直接的相互作用. 另外还有一种反应类型, P/N杂配铜基光催化剂除了参与光氧化还原催化循环之外, 自身还可以与底物发生相互作用, 如可以形成某种“底物与催化剂复合物”, 从而起到活化底物的作用, 事实上是发挥了电子转移和活化底物的双重催化功能, 我们把这一类反应归类于铜基光催化剂双功能催化的反应(Scheme 22), 下面予以介绍.
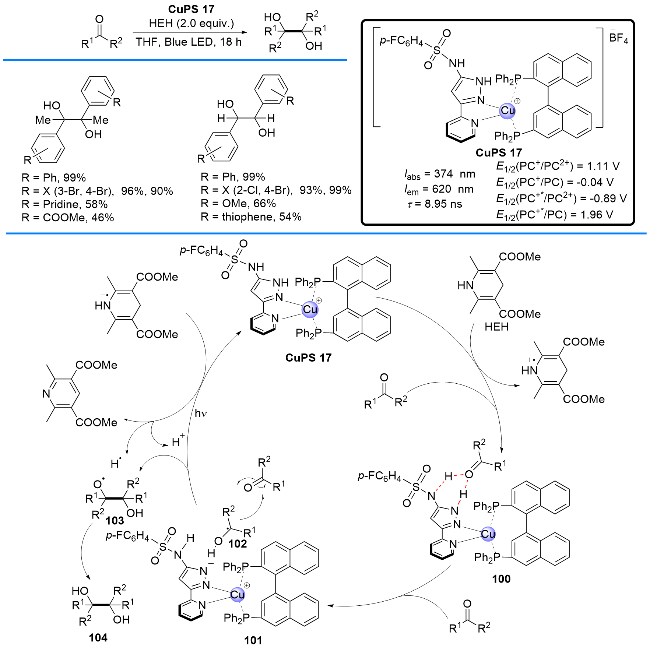
2019年, Collins课题组[39]开发了一种含有磺酰胺吡唑-吡啶配体的双功能P/N杂配铜基光催化剂, 并且利用该铜基光敏剂的双功能催化性能, 高效地实现了醛和酮的频哪醇偶联反应, 得到邻二醇类化合物(Scheme 23). 作者提出了可能的反应机理: 在光照辐射下, Cu(I)络合物首先转化为激发态Cu(I)*, 然后激发态Cu(I)*被汉斯酯还原淬灭形成Cu(0)状态100; 在该状态下, 磺酰胺吡唑-吡啶配体通过氢键形式活化醛(或酮), 这种质子偶合有利于Cu(0)把醛(或酮)还原成羰自由基物种102, 进而与另一分子醛(或酮)偶联形成氧基自由基中间体103; 最后, 103从汉斯酯自由基正离子物种转移一个氢原子, 从而形成产物邻二醇类化合物104.
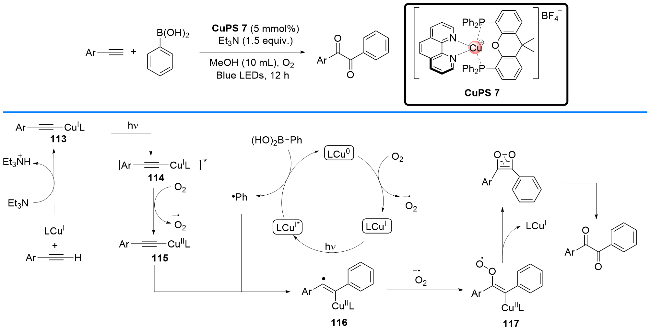
2021年, 于炜课题组[41]也报道了一例铜基络合物既充当光催化剂转移电子, 又充当过渡金属中心实现偶联片段偶联的反应. 他们以[Cu(DPEphos)(bcp)]PF6为光催化剂及过渡金属偶联中心完成了O-五氟苯甲酰基酮肟C(sp3)—H键的官能化反应, 成功地构建了3,4-二氢- 2H-吡咯类衍生物.
3 基于铜基光催化剂和其他金属的双金属协同催化反应
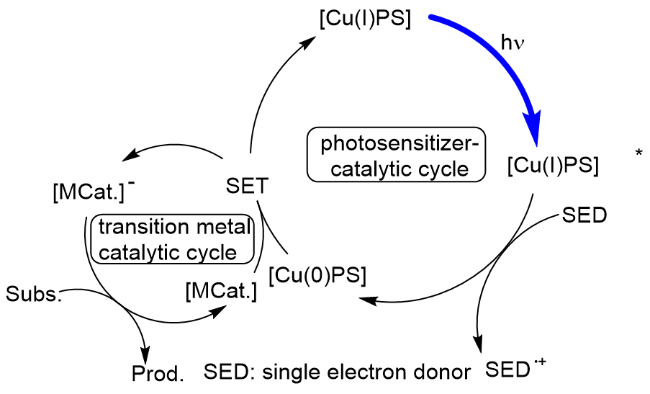
自从可见光驱动的光催化反应问世以来, 利用光催化剂光氧化还原与过渡金属催化相结合的光催化反应受到广泛关注, 并成为构建碳-碳键或碳-杂原子键的强力手段[1d,43]. 与光催化剂直接和底物作用的单电子转移机制不同, 这类反应的机制是光催化剂在可见光的照射下, 通过光氧化还原活化过渡金属催化剂, 然后底物进入到金属催化循环中, 从而生成预期的产物. 通常, 此种机制进行的光催化反应所使用的光催化剂主要是一些贵金属光催化剂, 如Ru(II)、Ir(III)光催化剂, 此外, 也有一些有机光催化剂, 如4-CzIPN等. 过渡金属催化剂则主要是一些镍、钴及铜等过渡金属配合物. 尽管铜基光催化剂在光催化反应中取得了长足进展, 但它们应用于和过渡金属相结合进行的双金属催化的光反应并不多见. 究其原因, 主要是铜基光催化剂中配体和中心金属铜的配位键不够稳定, 当有其他金属络合物存在时, 容易发生金属配体之间的相互冲突, 从而使得光催化剂容易改变结构而失活. 但是, 令人高兴的是, 在化学工作者不断地探索开发下, 已有一些报道介绍通过调控过渡金属与铜基光催化剂的配体, 使得两者能够协同催化反应. 我们把这一类反应归于铜基光催化剂和其他金属的双金属协同催化反应(Scheme 26), 下面予以介绍.
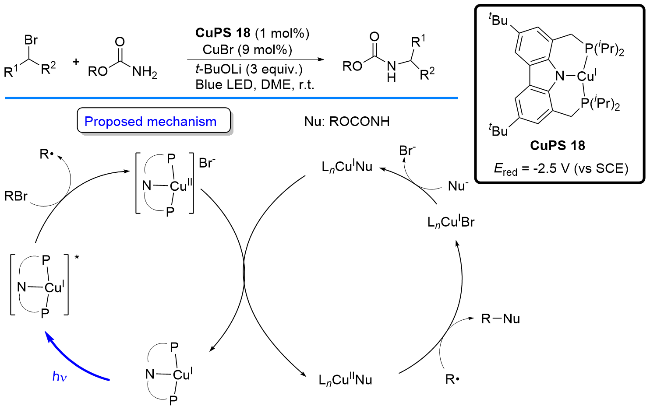
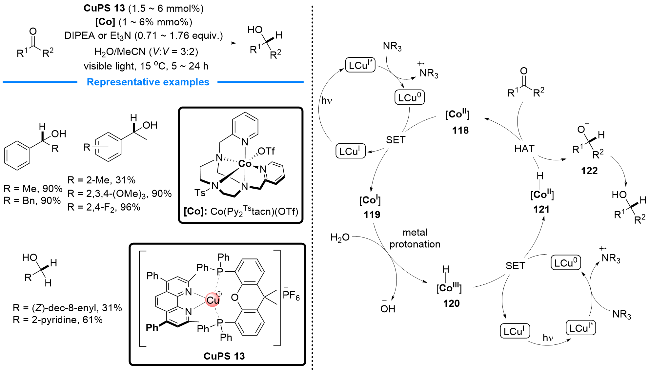
同年, Lloret-Fillol课题组[45]开发了一种钴络合物[Co(Py2Tstacn)(OTf)]OTf与P/N杂配Cu(I)光催化剂协同催化的醛、酮的氢化还原反应. 该反应直接以H2O和胺(NEt3或iPr2NEt)作为氢源(Scheme 28). 作者提出了可能的反应机理: 首先, 胺类物种被P/N杂配Cu(I)络合物光氧化成自由基阳离子并启动光催化循环; 接着Co(II)催化剂118被Cu(0)还原为Co(I) 119; 随后, 水提供氢质子并使Co(I)氧化生成H-Co(III)物种120, 接着120被Cu(0)还原为H-Co(II)物种121; 121与醛或酮发生亲核加成, 并质子化转化为醇类化合物, 释放Co(II)物种, 完成钴催化循环.
4 基于能量转移的催化反应
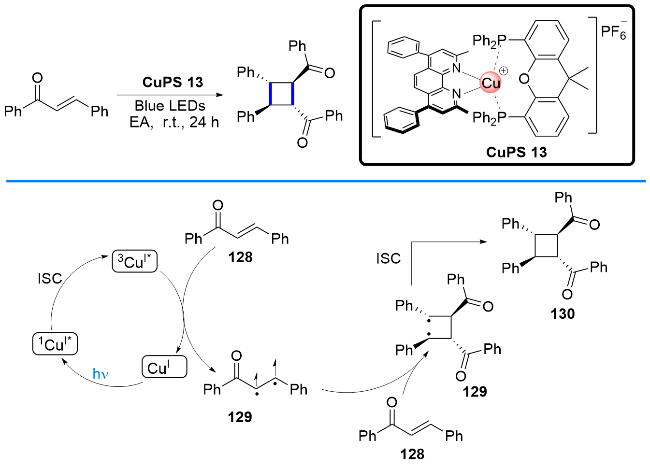
光催化能量转移(EnT)反应[47]是一种高效、绿色合成有机分子的光化学反应模式之一. 其原理是光敏剂(PS)被可见光激发, 较长时间处于高活性激发三重态(PS*), 从而可增强反应物对光照的吸收效率, 实现很多基态下无法进行的反应, 例如环加成、电环化反应、去外消旋化、迁移和重排等. 虽然通过能量转移机制成功地构建有机分子的光催化反应有不少报道, 但是所用到光敏剂主要涉及有机染料光敏剂, 如曙红Y和孟加拉玫瑰红等, 以及贵金属光敏剂, 如铱(III)和钌(II)光敏剂等. 而涉及铜基光敏剂参与的能量转移案例比较少见. 值得高兴的是, 近年来, 随着化学工作者们的不断研究探索, 以P/N杂配Cu(I)络合物为载体的能量转移光催化反应也逐渐被开发和报道.
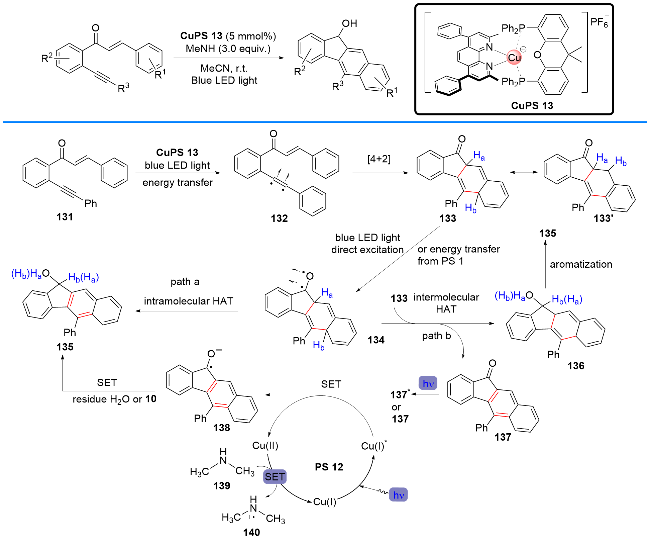
2021年, 刘运奎课题组[50]报道了一种在可见光照射和铜基光敏剂存在下, 将1,6-烯炔转化为苯并[b]芴醇的方法(Scheme 32). 值得一提的是, P/N杂配Cu(I)光敏剂在构建苯并[b]芴醇衍生物的过程中, 不仅充当了能量转移光催化剂, 同时还充当了光氧化还原催化剂. 这种同一光敏剂在一个反应过程中同时参与能量转移和单电子转移催化循环的例子是非常稀有的. 作者提出了可能的反应机理: 在可见光照射下, 1,6-烯炔131经能量转移得到三重态中间体132, 接着进行分子内[4+2]环化产生中间体133和133'; 随后, 133在光激发或经铜光敏剂能量转移产生三重态中间体134, 接着其经历分子内的氢原子转移(HAT)得到最终产物和苯并[b]芴酮135; 同时, 也存在着另一种机制(path b), 即134和133通过分子间氢原子转移产生中间体苯并[b]芴酮137, 再被铜光敏剂/NMe3催化体系还原成苯并[b]芴醇135.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 结论和展望
与主流的Ru(II)、Ir(III)等贵重金属光催化剂相比, P/N杂配铜(I)基光催化剂的开发和在光催化反应中的应用起步较晚, 即便如此, P/N杂配铜(I)基光催化剂在可见光驱动的光催化领域已经展示了其巨大的应用潜力. 与贵重金属光敏剂相比, 它们的中心金属铜在地球的储量更丰富、价格更具优势, 并且毒性较低. 此外, 有些P/N杂配铜(I)光催化剂在某些光催化反应中能够展示其独有的催化性能[27]. 再者, 鉴于这类光敏剂可以通过双磷和双氮配体的结构微调, 充分调控光敏剂的光物理和光化学性能, 从而对于开发出光催化性能更加优异的P/N杂配铜(I)基光催化剂充满期待. 从目前P/N杂配铜(I)光催化剂介导单电子转移、双功能催化、双金属协同催化及能量转移四个反应类型来看, 铜基光催化剂的结构是起关键作用的. 它们大都由双磷和双氮配体围绕一价金属铜中心形成一种类似“十字环扣”的刚性结构, 这种结构使得铜光催化剂能够拥有较长的激发态寿命[4]. 对于不同类型的反应, 铜光催化剂配体之间存在不同的电子和立体位阻环境, 从而使得不同的铜光催化剂具有不同的氧化还原电位及激发态能量等, 进而影响到各类反应的进行.
虽然P/N杂配铜(I)基光催化剂催化的反应取得了很大的进展, 但仍存在以下一些不足之处: 其一, 由于这类光催化剂配体与中心金属铜的配位键不够稳定, 在反应过程中容易转化为N/N均配的铜基光催化剂而失活, 导致催化剂的用量加大. 其二, 同样因为配体与金属铜结合不稳定的原因, 在双金属协同催化过程中容易与其他过渡金属络合物发生配体冲突而失活, 从而大大减少了该类光催化剂在双金属协同催化反应中的应用机会. 其三, 基于P/N杂配铜(I)基光催化剂催化的不对称光催化反应报道仍然偏少. 相信随着化学工作者对P/N杂配铜(I)基光催化剂的研究越来越深入, 以上不足之处一定会逐步解决, P/N杂配铜(I)基光催化剂也将迎来更大的发展.
(Zhao, C.)