


C—C键和C—X键的形成和转化是众多化学爱好者的研究课题. 过渡金属催化的交叉偶联反应是目前最成熟和不可或缺的构建C—C键和C—X键的工具之一. 早在1972年, Kumada等[1]报道了过渡金属催化有机卤化物与格氏试剂的交叉偶联反应, 这一发现引起了化学研究者的极大兴趣, 因为它开发了非常规键(碳卤键)的断开. 几年后, Mizoroki-Heck、Sonogashira-Hagihara、Negishi、Stille和Suzuki-Miyaura等[2]类型的研究方案被开发, 使得以芳基或乙烯基卤化物作为偶联试剂成为后来研究的首选, 并在制药和学术实验室得到广泛应用. 尽管芳基卤化物的使用已经深刻地改变了金属催化交叉偶联反应的格局, 但其在有机合成中的应用仍存在一些缺点: 卤化废物的形成、高度官能化的芳基碘化物和芳基溴化物的可利用性低等. 另外, 以化学和区域选择性方式获取有机卤化物时会遇到较大困难, 特别是在后期功能多样化的时候. 为此, 化学家们便开始寻找具有更灵活、通用和实用的芳基卤化物替代物. 考虑到芳基卤化物可由硝基芳烃的还原、重氮化和卤化获得, 若能实现硝基化合物和亲核试剂的交叉偶联, 则可以减少一步或多步步骤. 于是近年来开发了多种过渡金属催化硝基芳烃的脱硝基偶联反应[3]. 此外还开发了基于C—CN键断裂的芳香腈偶联反应[4]以及季铵盐C—N键断裂构建C—X键反应等[5].
在这些替代品中, 由于硝基的毒性较大、芳香腈的氰基易官能团化导致副产物增多等问题, 研究者便将注意力转移到苯酚衍生物, 因为它们是天然、丰富、无毒、易于获得和独特的反应中间体, 使得其在交叉偶联反应中具有较大的吸引力. 在过去的十几年里, 苯酚衍生物已被证明是芳基卤化物的一种可行且有力的替代物[6], 因氧原子与芳环存在p-π共轭, 苯酚的C—O键解离能(BDE)高达464 kJ/mol, 相比活泼的O—H键, 实现苯酚的C—O键活化显然较为困难[7]. 因此研究者设想通过引入保护基团从而弱化C—O键解离能, 芳基磺酸盐、芳基酯或芳基氨基甲酸酯衍生物等首先被实现过渡金属催化交叉偶联[8], 其机制一般包括形成Meisenheimer络合物或过渡金属插入形成氧化加成产物中间体, C—O键断裂反应速率如Scheme 1所示. Martin[9]、Li[10]、Lundberg[11]和曹志超[12]等已关于此部分做了详细的综述总结. 随着绿色化学发展的重要性日益显现, 因芳基磺酸盐的使用总会产生化学计量的含硫废物, 加上高昂的价格使得此类物质的应用前景受限.
芳甲醚(Ar—OMe)是一类基础且重要的原料和中间体, 也是苯酚的最简单衍生物, 在有机合成中常作为溶剂, 可用于生产香料、染料、医药和农药等. 通常这类化合物可由Williamson合成法或硫酸二甲酯与酚在碱性溶液中反应而轻松制备, 如今大多商业可购, 且价格低廉, 并且甲氧基的离去几乎不会对环境造成任何污染. 毫无疑问, 芳甲醚将成为芳基卤化物的替代物首选. 但C—OMe键具有较高的BDE (≈418 kJ/mol),[13] 甲氧基作为离去基团的能力较低, 其断裂活化一直是研究难点之一. 尽管与芳基卤化物或其他C—O亲电试剂的使用相比, 芳甲醚作为偶联剂的使用仍处于起步阶段, 但考虑到在苯酚分子中合成芳甲醚很容易, 可以预见的是, 通过C—OMe键作为一个反应位点转化为其他官能团在有机合成中必定具有重要研究意义.
在科学家的不懈努力下, 有关C—OMe键的活化已取得部分进展, 多种芳甲醚的C—OMe键官能团化也得以实现. 本文基于芳甲醚C(sp2)—OMe键的断裂, 按照不同反应原理(包括过渡金属催化的C—OMe键转化、自由基介导的C—OMe键转化、Brønsted酸或碱催化的C—OMe键转化)对近十年来关于芳甲醚转化反应进行综述, 探讨其相关反应机理和应用, 并对该领域的发展前景进行总结与展望.
1 过渡金属催化的C—OMe键转化
1.1 金属Ni催化的C—OMe键转化
1.1.1 氢化反应
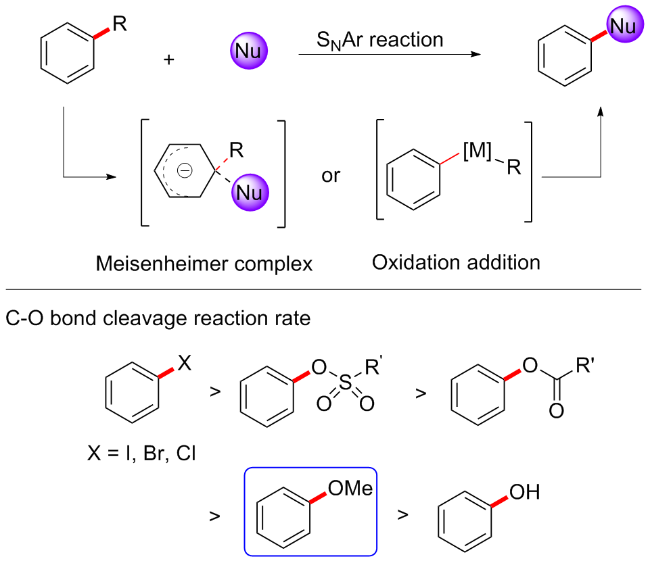
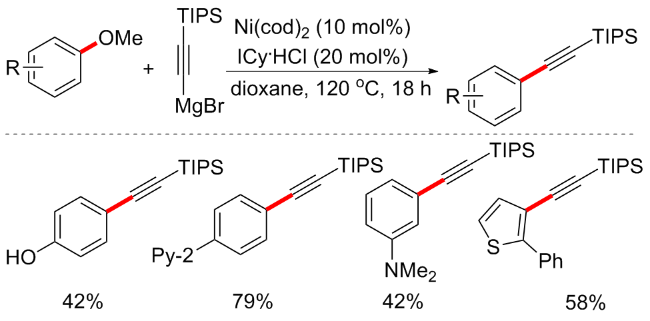
2015年, Chatani等[14]报道了一种镍催化的芳甲醚C—OMe键断裂氢化方案. 芳甲醚的C—O键还原裂解在有机合成中具有巨大的潜力, 虽然早期已经报道了几种可以促进芳基醚还原氢化的催化剂, 但这些催化体系均需引入外部还原剂, 例如氢硅烷或氢气等[15]. 在该方案中, 作者摆脱了外部还原剂的束缚, 以Ni(cod)2 (20 mol%)为催化剂, 连有金刚烷的N-杂环卡宾I(2-Ad)•HCl (20 mol%)为配体, 2 equiv. NaOtBu为碱, 在甲苯溶剂中加热至160 ℃反应18 h, 可实现芳甲醚的还原氢化(Scheme 2). 外部还原剂的缺乏使得催化体系需要特殊的配体参与, 作者通过筛选多种不同N-杂环卡宾配体, 最终发现Ni(cod)2/I(2-Ad)•HCl为最合适催化体系. 底物考察发现, 含缺电子取代基的芳甲醚表现较好, 含烷烃取代基的芳甲醚产率较低, 而具有供电子取代基的底物(如二甲氨基)不可耐受. 该方案不仅适用于芳甲醚, 含乙基、苄基、异丙基和苯基等取代的醚类均能在该体系中较好地还原氢化. 另外, 该方案的另一优势是, 因为不需要添加外部还原剂而具有独特的化学选择性, 即对烯烃和酮可选择性还原. 在探讨反应机制时, 作者通过和前述需要外部还原剂的机制一起分析, 用外部氢化物试剂还原裂解芳甲醚的机制可能涉及氧化加成形成镍(II)-甲醇盐A, 然后还原芳基-镍键(Scheme 2a); 用外部氢硅烷还原裂解芳甲醚的机制可能涉及生成甲硅烷基镍(I)中间体, 随后协同消除MeOSiR3 (Scheme 2b). 中间体B这种聚合物对于萘甲醚是较好的底物, 但并不适用于苯甲醚; 作者便猜想该方案应该是首先发生C—O键的氧化加成以形成中间体A, β-氢消除将导致中间体C的形成(Scheme 2c), 因此, 该方案能够在没有外部还原剂的情况下进行还原裂解, 但这一猜想作者并未通过实验证实.
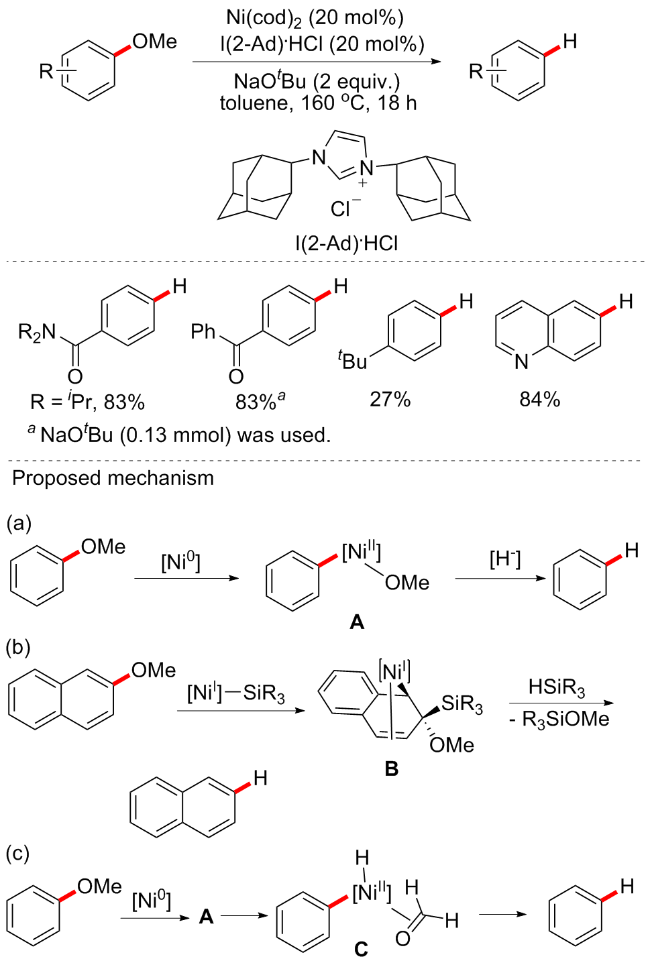
上述芳甲醚的还原氢化方案底物普适性欠佳, 随后该课题组[16]对芳甲醚的C—OMe键还原氢化进行了改良, 通过尝试8种不同还原剂, 包括HSiMe(OMe)2、HSiEt3、H2和9-硼双环[3.3.1]壬烷(二聚体) (9-BBN dimer)等, 最终发现二异丙基氨基硼烷是Ni(cod)2 (20 mol%)和卡宾配体(IMesMe 20 mol%)催化体系下的最适还原剂, 在甲苯溶剂中加热至160 ℃反应18 h, 可实现芳甲醚的还原氢化(Scheme 3). 虽然需要加入还原剂, 但该方案普适性非常好(55例子), 除了基本的缺电子取代基是良好的底物外, 烷烃(tBu)、四甲基硅烷(TMS)等以及含氮供电子基的吗啉、哌啶和哌嗪均可适用, 并具有良好的收率(产率48%~99%). 为了探究还原产物中氢化物的来源, 作者进行了一系列氘代标记实验. 但实验并不顺利, 氢化产物和甲苯溶剂之间也会发生H/D交换反应, 为了避免与溶剂的H/D交换, 当在1,4-二氧六环中进行氘代标记实验时, 芳甲醚中的C(Ar)—H键的H/D交换仍然阻碍了氢化物来源的探索. 尽管如此, 作者依旧认为氢化产物的氢来源于二异丙基氨基硼烷, 并提出的机制如下: 首先芳甲醚的氧原子与还原剂的硼原子配位以生成络合物A, 这可能是还原裂解的关键; 随后C—O键与Ni氧化加成, 形成中间体B; 随后的氢化物从硼迁移到镍, 然后以分子内方式通过C络合物形成氢化镍D, 最终通过伴随镍催化剂的再生和还原消除形成脱氧氢化产物E.
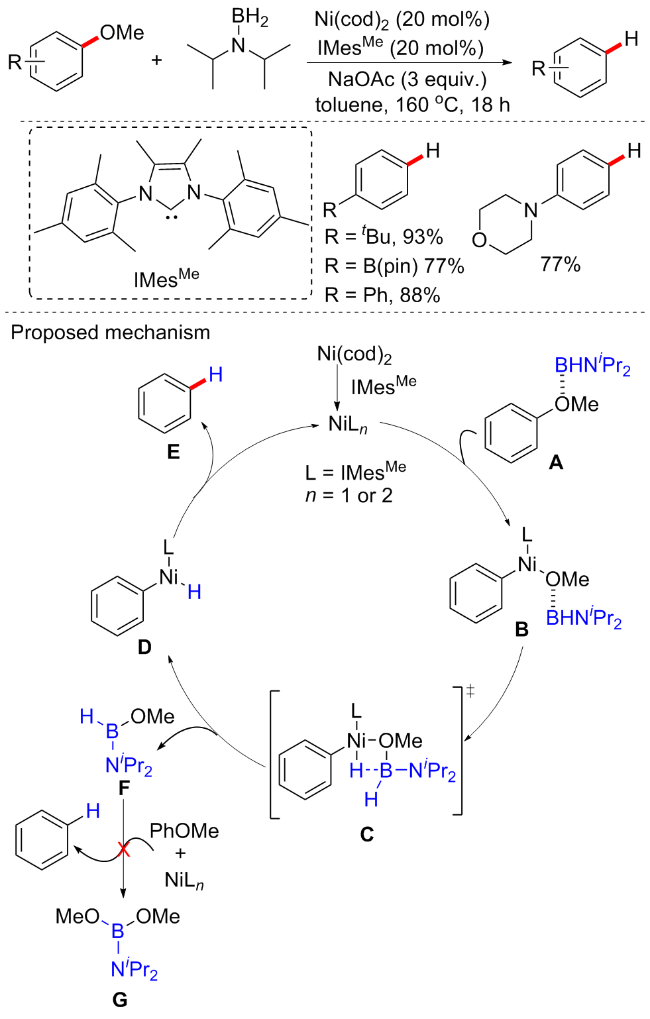
同年, 陈铁桥等[17]开发了一种在温和的反应条件下, 用易得的甲酸钠为还原氢化剂, 实现镍催化芳甲醚的高效氢化方案(Scheme 4). 该反应对于新戊酸萘酯类的C(Ar)—OPiv断裂可在微量的镍催化剂(低至0.5 mol%)下进行, 对于C—OMe键的断裂催化剂量仍需10 mol%, 但相较之前的20 mol%仍是一大进步. 并且, 此次经过氘化实验, 确认氢源自甲酸钠, 提出的机制如下: 首先, Ni插入C—O键形成氧化加成络合物A, 然后在甲酸钠作用下合成中间体B, 随后B的脱羧生成氢化镍中间体C. 最后还原消除产生氢化物并再生Ni(0)络合物. 早期Agapie等[18]还以(二膦)芳基甲基醚为基准物, 针对镍催化芳甲醚的C—OMe键还原氢解进行了详细机理研究.
1.1.2 烷基化反应
活泼的碳负离子(例如有机锂、格氏试剂等)作为直接的碳亲核试剂是最早被开发并被应用于有机合成的. 有机锂试剂由于高反应性和在偶联条件下的快速分解, 使得其应用于甲氧基取代偶联反应较为困难. 直到2014年, Rueping等[19]便以LiCH2TMS为亲核试剂, 实现了直接取代芳香族甲氧基的新合成途径, 并用于靶向和多样化的合成(Scheme 5a). 其实在先前的研究中发现, 膦配体对于辅助C—OMe键的活化是具有重要作用的[20], 但在该方案中, 在不需要PCy3(三环己基膦)等膦配体的条件下, 仅1 mol% Ni(cod)2便可以99%收率合成TMS取代的烷基化产物, 但底物大多局限于萘甲醚或是连芳基甲醚类衍生物(如4-苯基苯甲醚). 由于合成的ArCH2SiMe3是稳定、有活性的且易于进一步官能团化的中间体, 作者便以该物质为底物, 获得了许多其他重要底物, 包括烯烃、芳基β位的各种醇、胺、和芳香族羧酸等.
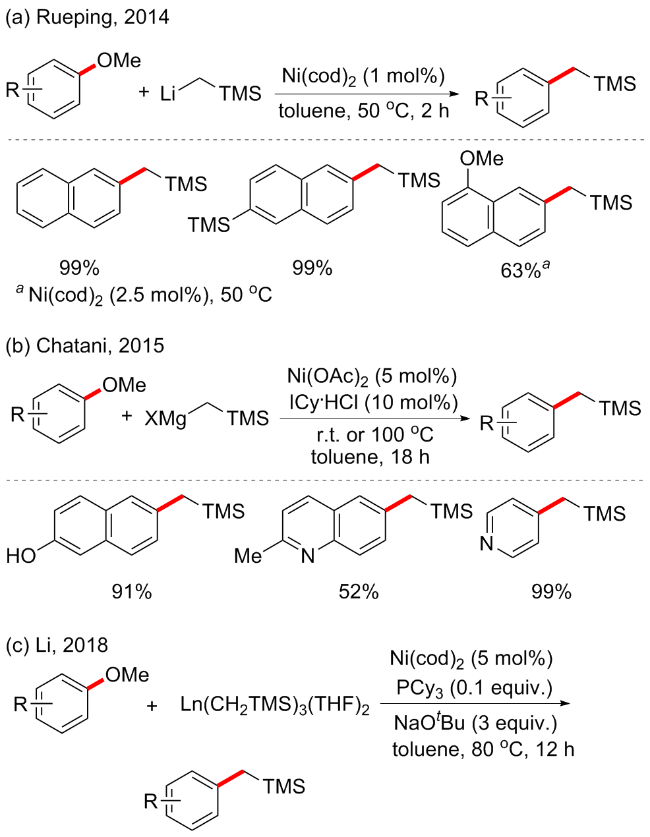
随后, Chatani等[21]开发了镍催化的烷基格氏试剂与芳甲醚的交叉偶联方案(Scheme 5b). 反应以N-杂环卡宾1,3-二环己基氯化咪唑(ICy•HCl, 10 mol%)作为配体, Ni(OAc)2 (5 mol%)为催化剂, 可合成多种TMS取代的烷基化产物. 该方案较上述普适性更广, 含活泼氢(如OH)的萘甲醚或者二甲氨基等供电子基以及杂环取代基均能在此条件下耐受. 对于格氏试剂, 除了Me3Si- CH2MgX, 还允许引入其他烷基类型基团, 包括Me、ArCH2、金刚烷基和环丙基等. 随后, 该课题组继续报道了以1,2-双(二环己基膦)乙烷(dcypt)为配体的镍催化烷基碘化镁和芳基醚的脱甲氧基偶联反应[22].
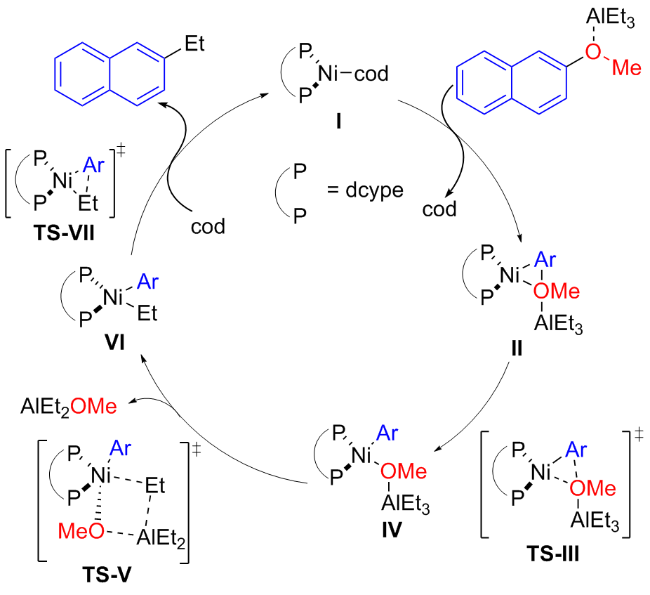
2015年, Chatani等[24]报道了Lewis酸(AlMe3)辅助镍催化的芳甲醚C—OMe键断裂的烷基化交叉偶联(Scheme 6a). 随后Rueping等[25]也开发了烷基取代的Lewis酸辅助镍催化的芳甲醚烷基化偶联(Scheme 6b). 该方案中, 芳甲醚衍生物可以有效地转化为各种烷基取代的化合物, 不受限于芳基醚的甲基化, 还可实现乙基化、异丙基化和环戊基化等, 含缺电子或供电子取代基的芳甲醚在该方案中也表现良好, 并具有中等至优异的产率(51%~98%), 但反应时间太长是该方案的缺点之一. 为了确认芳甲醚和三烷基铝之间的反应关系, 作者通过27Al和1H NMR光谱研究以及相关反应历程的计算研究, 表明芳甲醚C—OMe键的氧化加成受益于Lewis酸的配位, 即AlEt3与OMe基团的配位有利于NiII络合物IV的形成. 具体反应机制如Scheme 7所示.
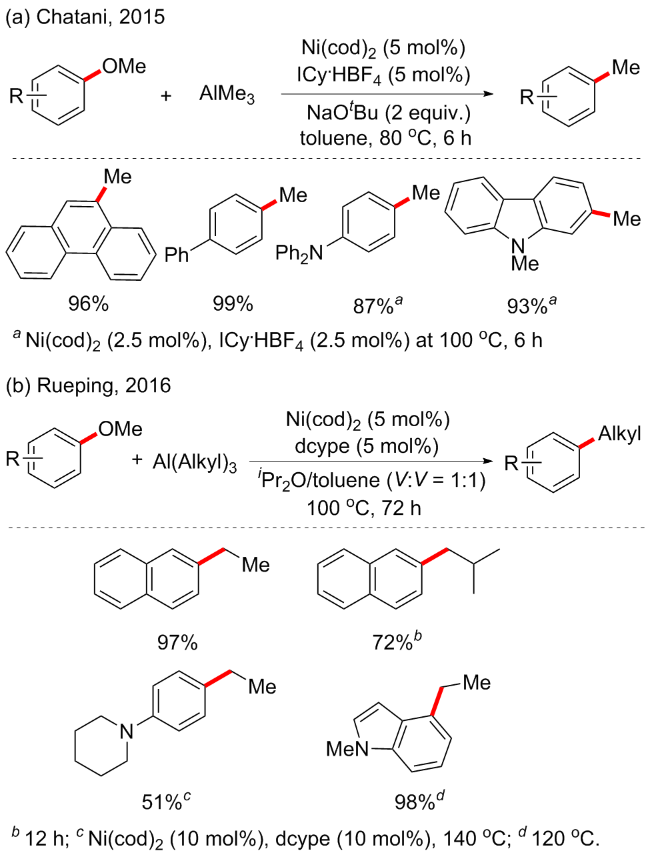
图式6 Lewis酸辅助镍催化的芳甲醚通过C—OMe键断裂的烷基化交叉偶联Scheme 6 Lewis acid assisted nickel-catalyzed alkylation cross- coupling of aryl methyl ethers by C—OMe bond cleavage |
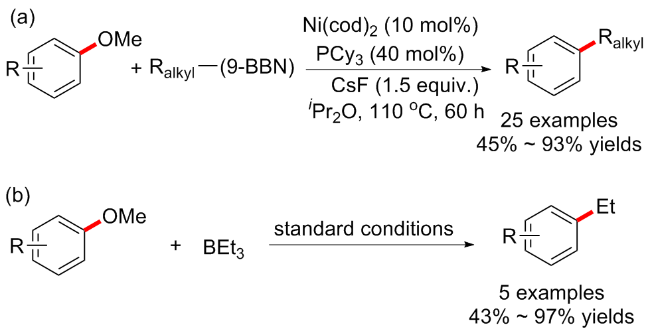
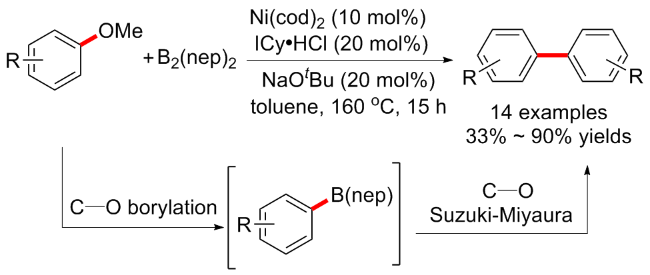
9-硼双环[3.3.1]壬烷(9-BBN)应用较为广泛, 通常用作选择性硼氢化试剂, 与烯烃反应有较高的选择性. 并且烷基硼烷由于其环境友好、操作简单和优异的官能团耐受性, 也被广泛用于天然产物和生物活性分子的合成[2,26]. 考虑到烷基硼烷可以充当路易斯酸(即与上述的三烷基铝有异曲同工之妙), 与烷氧基的配位应该对萘甲醚的氧化加成也有积极影响, 可能会克服一些不期望的副反应相关的限制, 包括经常观察到的硼氢化物消除等. 于是该课题组[27]继续开发了镍催化的芳基醚和烷基取代的9-BBN之间的交叉偶联(Scheme 8a). 在该反应条件中, 除了必要的金属镍和膦配体PCy3, 碱金属Cs对于反应的成功具有至关重要的作用. 当以CsF为添加剂时, 反应可以顺利进行, 而使用其他碱金属添加剂时则没有反应. 该方案底物普适性良好, 产率高, 但反应时间较长(60 h). 有趣的是, 除了芳甲醚外, 烯醇醚类的衍生物也耐受, 并且反应时间缩短至18 h. 最后, 作者还发现带有各种官能团的多环芳香甲基醚在标准条件下也可以良好的产率与三乙基硼烷交叉偶联(5例子, 产率43%~97%) (Scheme 8b).
1.1.3 芳基化反应
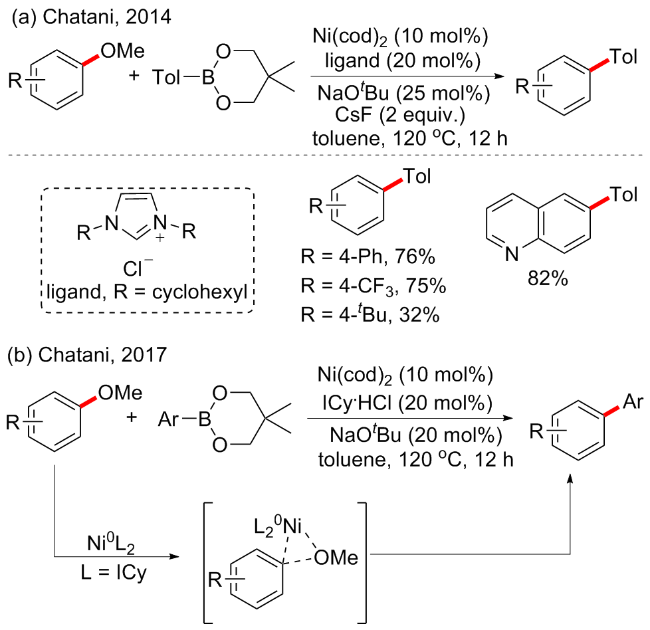
提及芳基化偶联, 或许最先想到的莫过于铃木反应(Suzuki-Miyaura reaction). 当研究者尝试将甲基醚作为潜在的亲电试剂替代卤素时, 由于甲基醚的C—OMe键富电性很强, 使得它通过氧化加成进行活化具有一定挑战性, 特别是和有机硼烷化合物反应时, 这一因素限制了它在交叉偶联反应中的应用. 在2014年, Chatani课题组[28]开发了镍催化的芳甲醚与有机硼试剂的交叉偶联方案(Scheme 9a). 该反应的成功归因于氮杂环卡宾(NHC)配体(ICy)的使用, 作者通过筛查17种不同NHC配体, 最终发现当R=cyclohexyl(环己基)时, 表现最好, 该配体也促进一系列以前未成功的芳甲醚Suzuki- Miyaura型交叉偶联反应, 但在该反应中缺电子取代基底物相比供电子取代基表现更好, 含烷烃的芳基醚不反应或者产率较低. 其实早在2008年, 该课题组[29]就报道了镍/PCy3催化的甲氧基芳烃与芳基硼酸酯的交叉偶联, 其中化学计量的碱金属添加剂如CsF的加入对反应的进行至关重要, 但镍/ICy催化的交叉偶联不需要添加化学计量以外的生物碱. 随后在2017年, 该课题组[30]针对这几种不同催化体系催化芳甲醚与有机硼烷偶联的反应机制进行了大量的理论和实验分析, 包括密度泛函理论等(Scheme 9b), 研究表明在镍/PCy3催化反应中, C—OMe键氧化加成的活化能太高, 在催化条件下无法发生, 但当CsF和芳基硼酸酯与Ni(PCy3)2/甲氧基芳烃片段相互作用形成四元络合物时, 氧化加成过程在能量上变得可行, 而在镍/ICy催化的反应中, C—OMe键的氧化加成不需要CsF的帮助, 因为镍配体键更强, 稳定了过渡态. 随后, Ar—Ni—OMe中间体的催化被确定为通过能量低于β-氢消除所需能量的途径进行, 反应的总驱动力是形成碳碳键的还原消除.
同年, 孙宏枚等[32]开发了一种新的NHC/膦镍配合物制备方法. 由于Ni(II)配合物[NEt4][Ni(PPh3)X3] (X=Cl和Br)中的季铵阳离子可以在[NEt4]-Ni(PPh3)X3]与大体积ItBu (1,3-二丁基咪唑-2-亚基)或IPr [6-二异丙基苯基)咪唑-2-亚乙基]配体的反应中充当良好的离去基团, 可容易地被膦配体(PCy3)取代, 便可以高产率得到相应的混合NHC/膦镍配合物(Scheme 11a). 在芳基格氏试剂与多种亲电试剂的交叉偶联中, 作者研究了它们对改变卡宾配体(ItBu vs IPr)以及膦配体(PPh3 vs PCy3)的催化行为. 除了对其催化活性有显著的协同作用外, 基于合理的结构设计, 实现了C—Cl、C—F和C—O键活化和转化的高选择性. 其中, 配合物Cat.1显示了芳甲醚与格氏试剂交叉偶联反应的巨大潜力, 产率优异(89%~97%). 但总体底物普适性一般, 芳甲醚大多局限于萘环或联苯基衍生物, 而格氏试剂则限于供电子或烷烃取代基等底物.
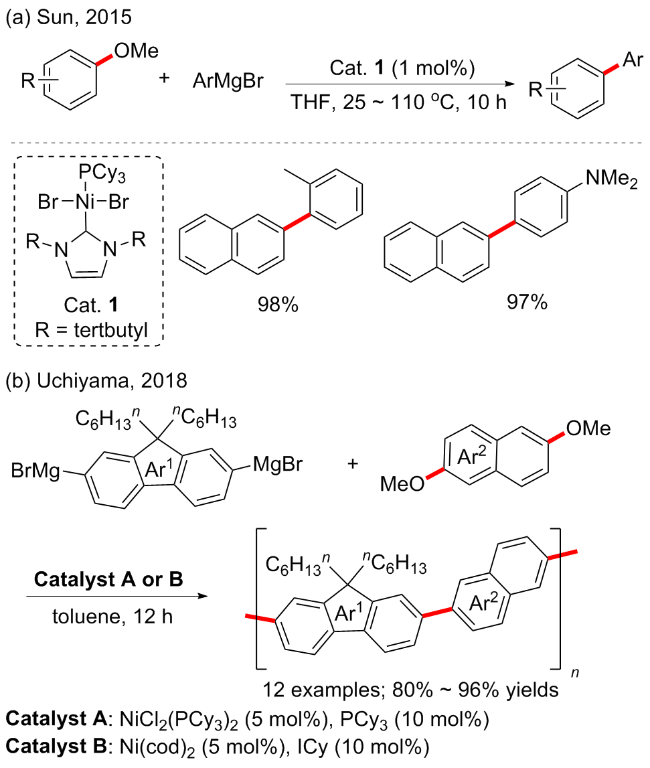
π-共轭聚合物广泛应用于光电子领域, 用于制造有机光伏器件、有机发光二极管和有机场效应晶体管等. 2018年, Uchiyama等[33]开发了一种通过芳甲醚C—O键断裂合成π-共轭聚合物的有效方法. 通过使用市售的镍催化剂, 在温和的条件下, 格氏试剂或有机锂试剂和醚之间的交叉偶联缩聚可顺利进行, 以良好至优异的产率提供了高分子量的缩聚产物(Scheme 11b). 有机金属偶联剂的纯度对于该反应具有重要影响. 该方法适用于在其他聚合过程中不反应的单体, 并为将一系列天然丰富的化学物质转化为有用的功能性化合物/聚合物提供了可能性.
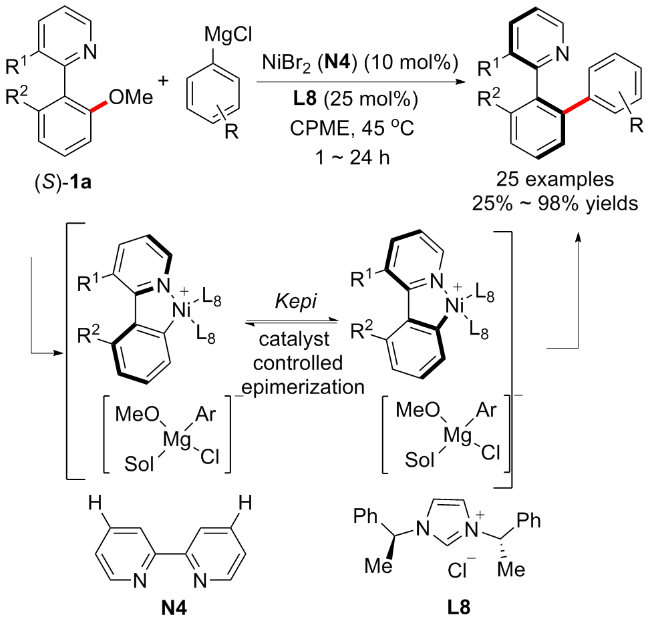
最近, 曹志超等[34]开发了一种通过镍催化C(Ar)—OMe键的动态动力学不对称交叉偶联实现苯甲醚衍生物对映转化的方案, 成功地构建了多种轴向手性杂环 (Scheme 12). 在该方案中, 合适的配体对镍催化反应至关重要, 由于膦配体的不适当电子和空间效应, 总是得到外消旋体或低收率对映体. 鉴于手性NHC配体在金属催化的非反应性化学键的不对称切割中诱导对映体选择性的良好能力, 在考察了多种手性NHC配体后, 最终发现L8为最优配体. 反应具有良好的普适性和产率, 含氮基序的杂二芳基甲醚, 包括异喹啉、菲啶和吡啶均适用于对映选择性交叉偶联. 由于外消旋杂二芳基甲基醚的两种对映体都可以转化为目标产物的相同对映体, 因此镍催化的苯甲醚衍生物对映选择性交叉偶联本质上是一个对映转化过程. 为了确定对映体转化是通过C(Ar)—OMe键的动态动力学不对称交叉偶联而不是其他途径实现的, 作者在45 ℃的环戊基甲基醚(CPME)中搅拌化合物(S)-1a, 进行了不同反应时间的消旋化研究, 观察到(S)-1a的缓慢外消旋, 从而表明对映体选择性交叉偶联不太可能通过由底物外消旋启动的常规动态动力学拆分途径发生. 机理研究以及密度泛函理论(DFT)计算表明, 这种转化的对映体转换可能是通过手性配体控制的非对映五元氮杂镍环物种的外嵌合, 而不是由传统的动态动力学解析实现.
Uchiyama等[35]在2016年还报道了镍催化的芳基醚和有机锂试剂的交叉偶联反应, 并具有良好的底物普适性和产率(Scheme 13). 此外, 关于镍催化的有机金属试剂和芳基醚的交叉偶联反应在早期也被报道过[36]. 尽管镍催化的芳基醚与有机锂试剂的交叉偶联已得到充分证实, 并成为苯酚衍生物转化为功能化芳香分子的有力合成工具之一, 但作用机制仍没有得到充分阐明. 最近, Payard等[37]在DFT计算的基础上, 通过与光谱和动力学数据的比较, 结合关键有机金属中间体的分离, 研究了Ni(COD)2催化芳香醚与PhLi的交叉偶联反应机理. 作者以2-甲氧基萘为模型底物, 首先将两个PhLi与Ni(COD)2进行络合, 形成富电子的镍酸锂[Li2(THF)2- Ph2Ni(COD)]; 芳香醚可以通过Li…O相互作用进行配位, 通过萘环与Ni的π配位促进剩余COD配体的置换; 随后共配合物LiOMe与PhLi交换, 然后还原消除以提供交叉偶联产物2-苯基萘, 并再生Ni0-酸锂配合物, 完成催化循环. 实验研究已经确定了镍酸锂的关键物种, 并阐述了溶剂在反应中的重要作用, 也揭示了锂在反应中的多重作用.
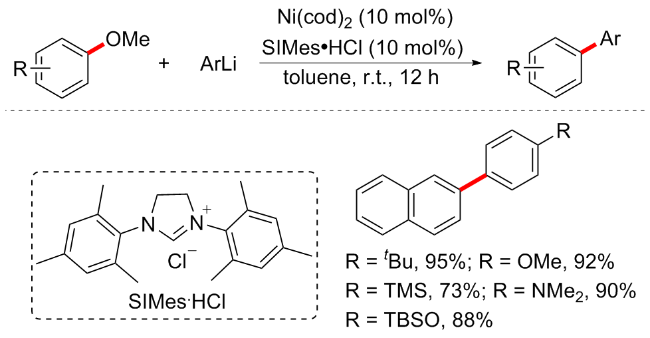
2016年, 施章杰等[38]开发了在镁作为金属还原剂的条件下, 芳基醚和芳基溴化物之间通过镍催化的亲电交叉偶联反应(Scheme 14). 反应条件温和, 底物普适性较为广阔, 多种供电子取代基的芳基溴能以很好的产率合成芳基化产物, 但缺电子基溴苯不可耐受. 对于芳甲醚而言, 依旧局限于大π环体系包括萘环或者联苯类的芳烃. 此外, C(sp3)—OMe键的活化也适用于该条件, 并能保持很好的ee值. 因为芳基醚既可与芳基硼酸偶联也可与溴苯偶联, 所以这一机理显得较为特殊. 一系列的对照实验表明, 这种交叉偶联并不是格氏试剂(溴苯和镁在该条件下形成苯基溴化镁)形成的简单组合; 此外, 汞中毒试验结果表明, 催化循环中可能涉及多相物种; 通过添加2,2,6,6-四甲基哌啶氧化物(TEMPO)等自由基抑制剂后芳基化产物未能观察到, 表明自由基物种可能参与催化循环, 但自由基钟试验却未能获得目标产物. 所以该反应机制并未被研究清楚, 有待进一步探索.
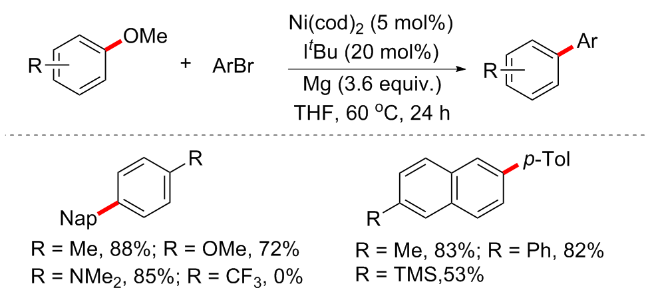
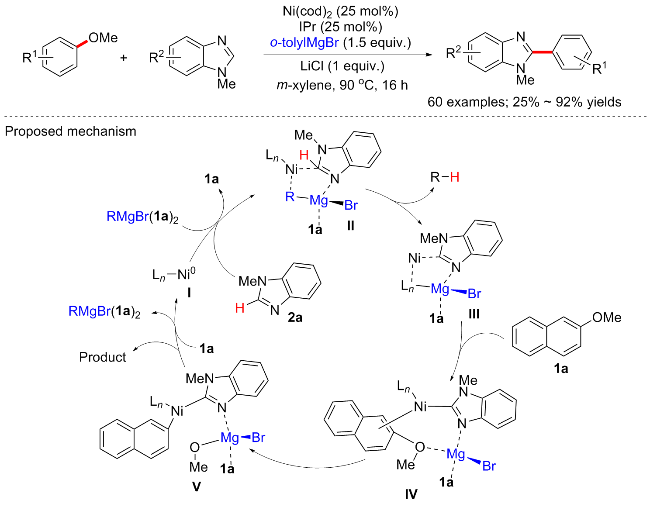
格氏试剂不仅能作为亲核试剂, 也可以作为芳基醚偶联的反应促进剂. 2018年, Ong等[39]实现了镍催化的芳甲醚与杂芳烃的C—H键活化偶联反应(Scheme 15). 反应以Ni(cod)2 (25 mol%)/IPr (25 mol%)为催化体系, 邻甲基苯基溴化镁和氯化锂为添加剂, 在90 ℃下反应16 h, 得到C—OMe键断裂的芳杂环偶联产物, 有趣的是, 该方案并不会产生上述的芳基醚与格氏试剂偶联的Kumuda型偶联产物. 该反应适用于广泛的底物, 包括萘基甲醚、茴香醚和多种其他杂芳烃衍生物(60例子, 产率25%~92%). 实验表明, 同时激活C—H/C—O键的关键是使用空间要求较高的邻甲苯基溴化镁. 详细的机理研究表明, 镍促进的1-甲基苯并咪唑2a与过量的RMgBr的脱质子化反应产生中间体II, 中间体II经历C—H活化步骤, 生成络合物III. 2-甲氧基萘1a与III结合将产生Ni络合物IV, 随后可通过氧化加成步骤进一步转化为中间体V. 重要的是, RMgBr和镍之间的协同作用对降低C—O/C—H裂解的活化能势垒至关重要. 最后, 络合物V的还原消除完成催化循环, 以提供所需的目标产物. DFT计算也支持了这一过程.
1.1.4 其他官能团化反应
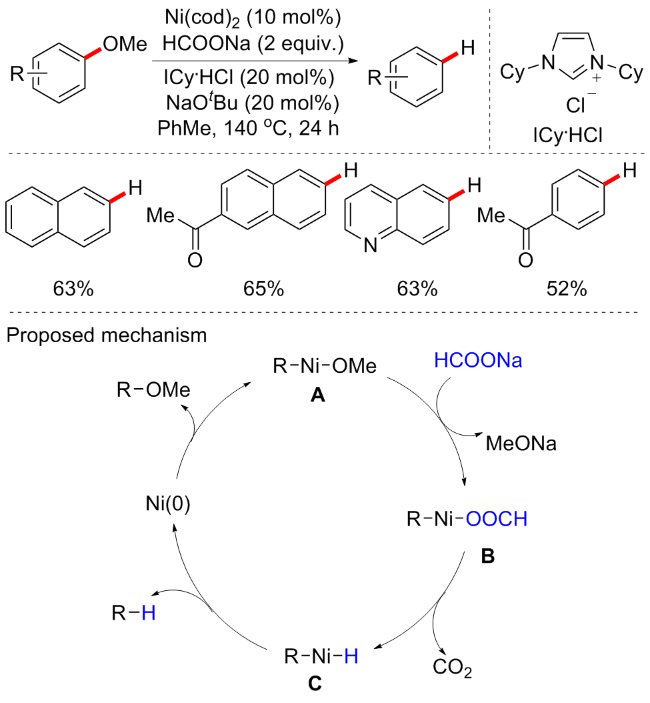
过渡金属催化形成碳-杂键的转化在有机合成中至关重要. 尽管芳甲醚作为偶联试剂具有巨大的潜力, 但通过C—OMe裂解形成金属催化的C—X原子键的情况非常罕见. 在过去的几十年里, 有机硼试剂作为关键的合成中间体在工业化学和有机合成一直备受关注, 以高效的方法合成有机硼烷试剂具有重要的意义. 近年来已实现了部分合成它们的催化方法, 例如Martin等[40]开发了镍催化的芳基醚ispo-硼烷化反应(Scheme 16a). 通过筛选多种金属和配体, 最终发现依旧是Ni(cod)2和膦配体的催化体系最优, 并需要3 equiv. HCOONa的辅助催化, 可以88%的收率合成硼烷化产物. 底物普适性良好, 具有邻位酯、三氟甲基或酰胺的各种芳甲醚都可以很好地被耐受, 很好地说明了该方案的化学选择性. 关于硼烷化的机理未在文中讨论.
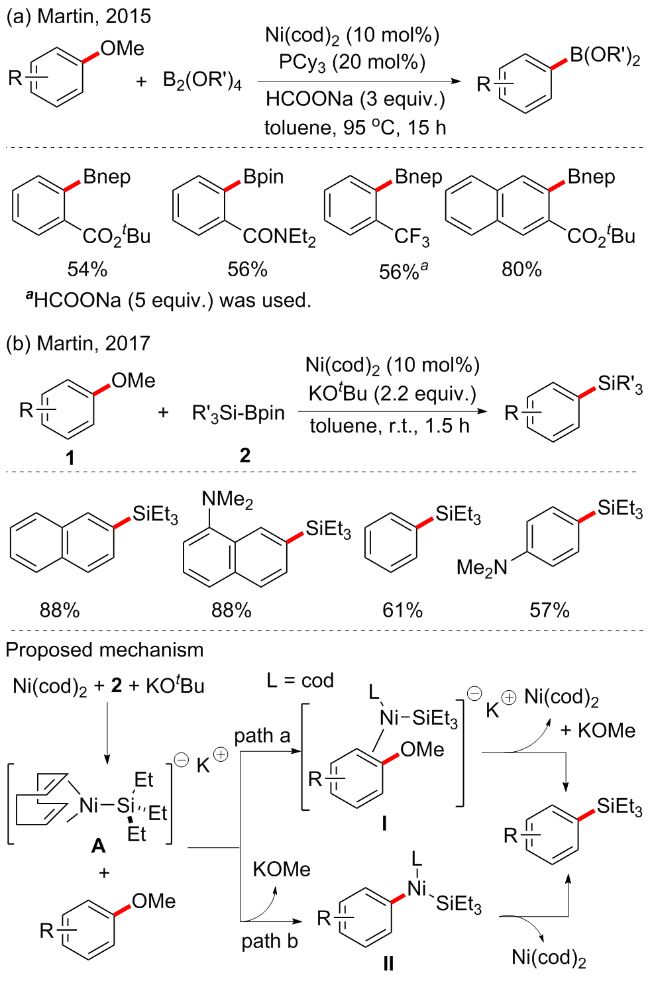
随后, 在上述芳基醚的硼烷化反应的思路下, 作者尝试将二硼烷试剂替换为烷基硅基取代的硼烷, 继续实现了镍催化的芳基醚硅烷化反应(Scheme 16b)[41]. 在温和条件下, 通过不依赖外部配体的情况下实现了镍催化的甲硅烷基化反应, 这代表了通过C—OMe断裂设计新的碳-杂原子键形成的重要一步. 底物普适性优越, 对于无取代的苯甲醚也适用. 文中还描述了该变换在正交甲硅烷化技术以及进一步衍生化中的应用. 初步的机理实验表明, 该机制的关键催化中间体(A)可能由Ni(cod)2、2和KOtBu原位生成. 尽管进行了大量实验, 但没有分离出A或得到直接的光谱数据, 可能是由于该中间体异常灵敏和寿命有限. 作者认为硅烷化的反应机制可能由以下两个途径进行: 包括金属催化的芳香亲核取代过程(path a), 和C—OMe键的氧化加成过程(path b).
1.2 金属Ru介导/催化的C—OMe键转化
1.2.1 氢化反应
2018年, Snieckus等[43]开发了一种新的钌催化的邻甲氧基苯甲酰胺脱甲基氢化反应, 该反应涉及以酰胺基团为导向的C—OMe键活化和氢化物还原(Scheme 18a). 最优条件是在RuH2(CO)(PPh3)3催化下进行, 使用三乙基硅烷(Et3SiH)或二异丁基氢化铝(DIBAL-H)作为还原剂, 在甲苯溶剂中125~135 ℃下反应20 h. 反应普适性良好, 对于包含多个甲氧基的化合物具有良好的位置选择性, 也是对镍催化的芳基醚脱甲氧基氢化方案的一个补充.
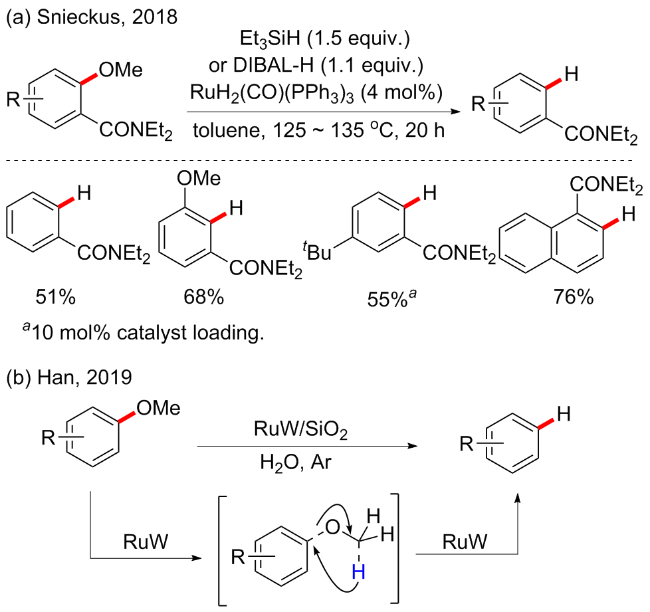
随后, 韩布兴等[44]提出了一种芳香醚的自我支撑氢解(SSH)方案, 通过使用反应物内的氢源(OCH3)产生芳烃, 并发现RuW合金纳米颗粒是反应的有效催化剂(Scheme 18b). 与以往报道的C—OMe键裂解氢化不同, 该方法的独特之处在于不需要外源氢或其他还原剂, 可以完全避免芳香环的氢化. 在醚完全转化时, 对芳烃的选择性可以达到>99.9%. 此外, 木质素也可以在RuW合金催化剂上有效地转化为芳烃. 实验结果和DFT计算证实了RuW催化剂中相邻的Ru和W物种协同作用以加速SSH反应.
1.2.2 芳基化反应
巧妙地理解和运用导向基团将大大加快一些复杂化学键功能化的发现. 随着研究的发展, 多种官能团, 如酰胺、亚胺、酮、羧酸、酯和杂环等已被开发并确定为位点选择性官能团化的强大导向基团[45]. 2015年, Snieckus等[46]开发了以酰胺为导向基团, 通过甲氧基断裂的邻甲氧基芳甲酰胺与芳基硼酸酯的交叉偶联来合成双芳基、杂双芳基和聚芳基分子的方法(Scheme 19a). 该反应基于以RuH2(CO)(PPh3)3为催化条件, 通过邻近的酰胺导向基团激活未反应的C—OMe键. 由此建立了一种无碱参与的偶联方案. 此外, 酰胺导向基团活化C—OMe是高度区域选择性的, 没有竞争性C—H活化过程干扰, 并提供了与乙酰基或新戊酰基相比更易于合成操作的酰胺功能性产物. 底物普适性良好, 产率中等至优异(40例子, 产率30%~99%). 实验研究表明, 异构体1-甲氧基-2-萘酰胺和2-甲氧基-1-萘酰胺可以优异的产率提供一系列芳基化萘酰胺产物, 并且远大于3-甲氧基-2-萘甲酰胺的反应活性. 随后该课题组继续报道了在同样催化条件下, 以羧酸酯为导向基团的邻甲氧基苯甲酸甲酯和芳基硼酸的交叉偶联方案(Scheme 19b), 并同样具有良好的底物普适性(25例子, 产率30%~99%)[47].
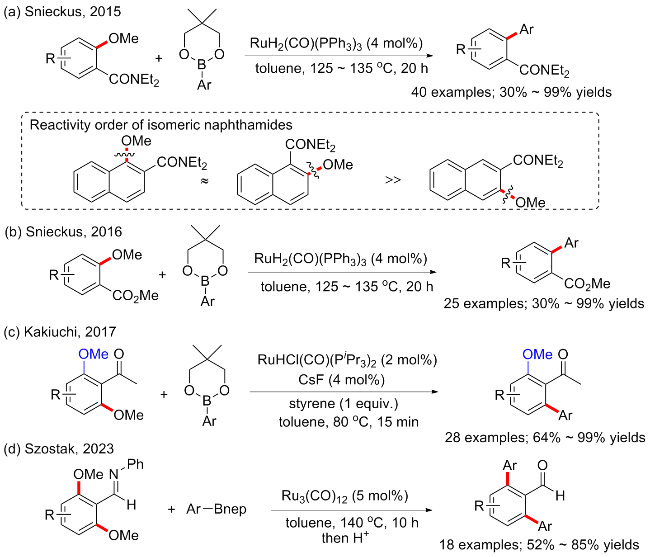
2017年, Kakiuchi等[48]报道了以乙酮基为导向基团, 钌催化的带有两个邻位C—OMe键的芳香酮选择性单芳基化方案(Scheme 19c).由RuHCl(CO)(PiPr3)2、CsF和苯乙烯组成的催化剂体系下, 2′,6′-二甲氧基苯乙酮与苯基硼酸酯的C—O芳基化反应选择性地生成C—O单芳基化产物. 选择性C—O单芳基化适用于各种芳基硼酸酯和芳香酮, 并具有较高的区域和化学选择性, 而当以RuH2(CO)(PPh3)3为催化剂时, 则生成双芳基化产物, 无选择性[49].
最近, Szostak等[50]还报道了以亚胺为导向基团, 钌催化的芳基醚和芳基硼酸酯偶联方案(Scheme 19d). 该方法以Ru3(CO)12 (5 mol%)为催化剂, 无需碱和配体, 可使苯甲醚快速合成有价值的二芳醛化合物.
1.3 金属Cr催化的C—OMe键转化
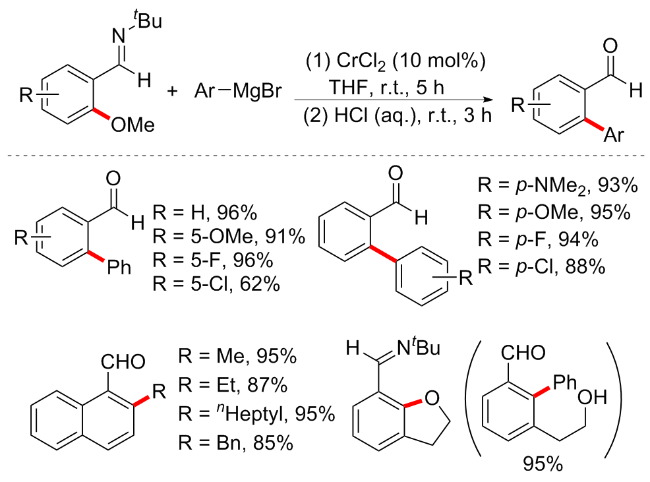
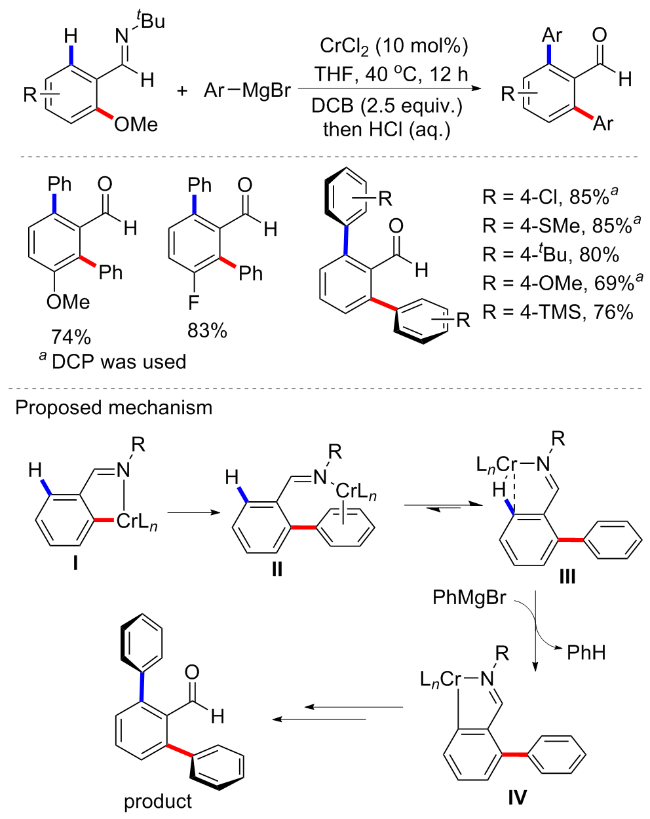
通过C—O键断裂实现芳基醚的偶联反应已被视为一种高效原子经济型的方法, 但以往主要局限于过渡金属镍催化, 而当多个C—OMe键同时存在于同一分子中时, 选择性催化一直是该领域的一个难点. 因此导向基团的引入对于C—OMe键的选择性活化是个较大的突破, 尽管已经开发了较多的钌介导/催化的C—OMe键转化方案, 但较为昂贵的RuH2(CO)(PPh3)3催化剂使得其实用性有限. 2015年, 曾小明课题组[51]利用廉价的铬催化芳基醚与格氏试剂通过C—OMe键断裂来实现选择性交叉偶联反应(Scheme 20). 在室温下, 使用简单、廉价的铬(II)催化剂, 结合亚氨基作为导向基团, 实现了多种转化. 该方案无需配体参与, 在CrCl2 (10 mol%)催化下室温反应5 h, 随后在盐酸作用下水解即可得到邻芳基取代的芳基醛衍生物. 具有非常良好的底物普适性, 含有供电子、缺电子等取代基的芳基醚或格氏试剂均能在此条件下实现高效率的偶联. 有趣的是, 铬催化的方案在亚胺基的辅助下也显示出2,3-二氢苯并呋喃的高效率开环/交叉偶联转化, 以95%的产率产生羟基连接的二苯甲醛. 对照实验显示, 用有机锌试剂Ph2Zn代替格氏试剂导致偶联转化率降低, 说明与有机镁相比, 有机锌的还原率更低. 此外, C—OMe键的交叉偶联反应效率受到芳香醚的电子效应影响, 缺电子取代的芳基转化效率高于供电子基芳基醚.
随后, 该课题组[52]在上述催化条件下, 通过添加1,2-二氯丙烷(DCP)或1,2-二氯丁烷(DCB)作为添加剂, 不仅实现了铬催化的芳基醚与格氏试剂交叉偶联, 还可实现邻位C—H键的活化, 从而将两个相同的芳基引入苯甲醛的邻位(Scheme 21). 有趣的是, 既然DCB或DCP是铬催化以亚胺基团为导向的邻位C—H键活化的关键, 那么如果第一步不添加配体, 先在Scheme 21的条件下实现C—OMe键的活化, 接着通过添加DCB实现邻位C—H键的活化. 基于此思路, 作者实现了将不同的芳基引入苯甲醛的邻位. 该方案同样具有良好的底物普适性和产率, 也适用于2,3-二氢苯并呋喃衍生物的开环活化偶联. 作者猜测在邻位C—O键官能化之后, 低价Cr可能与亚胺基团和结合的芳烃基团(中间体II)发生配位, 但详细机理并未在文中讨论.
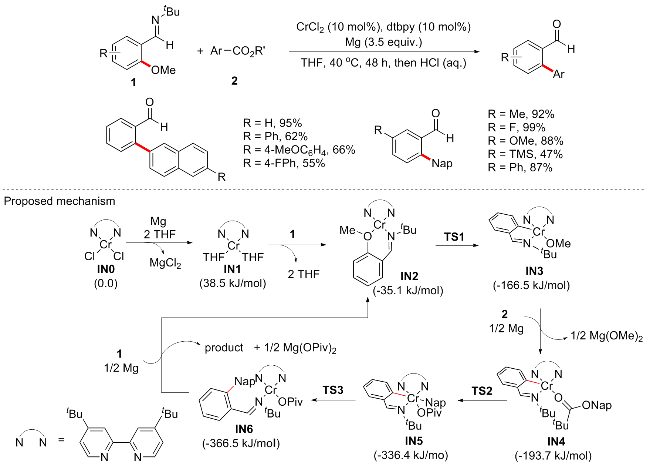
2020年, 该课题组[53]继续开发了铬催化的芳基甲醚衍生物与芳基酯在温和条件下的交叉偶联反应(Scheme 22). 作者认为两个芳基亲电试剂的还原交叉偶联通常涉及两个氧化加成过程, 此前已证明处于低氧化状态的反应性过渡金属Cr有利于C—OMe键的氧化加成活化. 作者首先以邻亚胺苯甲醚和芳基羧酸酯为底物对其进行条件优化, 通过筛选催化剂CrCl2、FeCl2、CoCl2和Ni(cod)2等, 依旧是低价CrCl2最优, 其他过渡金属无法催化. 最后以CrCl2 (10 mol%)、dtbpy (10 mol%)和Mg (3.5 equiv.)为催化体系, 实现了芳基醚和芳基羧酸酯的还原交叉偶联. 这一反应使得两个不同的未活化的C—OMe和C—C键通过化学键的同步裂解实现以前难以实现的转化. 底物普适性良好, 含供电子和缺电子取代基的芳基醚或羧酸酯均能在此反应中耐受. 根据控制实验和DFT计算, 作者提出的机制可能如下: 五重基态的Cr(0)络合物IN1 (38.5 kJ/mol)首先通过Mg还原形成, 分别位于相应的低自旋态和中等自旋态(单线态和三线态)之下238.1和97.9 kJ/mol. 底物1向IN1靠近并通过配体交换产生IN2 (-35.1 kJ/mol). 通过TS1 (-10.9 kJ/mol)过程促进o-C—O键与Cr的氧化加成, 从而提供IN3 (-166.5 kJ/mol). 随后, 继续在Mg的还原作用下, 通过TS2 (-105.0 kJ/mol)过程实现底物2的氧化加成, 提供IN5 (-336.4 kJ/mol). 最后, Cr的还原消除使C—C键形成, 产生IN6 (-366.5 kJ/mol), 然后还原以完成催化循环.
1.4 其他过渡金属催化的C—OMe键转化
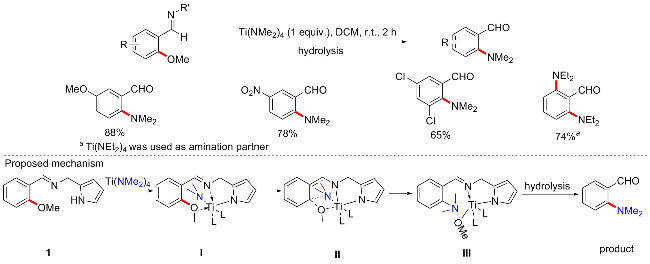
芳香胺在医药、农药、染料和聚合物工业中都具有重要作用. 传统上, 芳基胺可通过芳香硝基化合物的金属催化氢化制备, 如今更多的是通过金属催化偶联反应构建C—N键, 包括Buchwald-Hartwig、Ullmann和Chan-Lam等反应. 2015年, 李亚红等[54]开发了一种通过亚胺导向的Ti(IV)催化的芳甲醚和Ti(NR2)4之间的芳甲醚胺化方法(Scheme 23). 该方案条件温和, 操作简单, 无需配体参与, 在室温条件下即可实现亚胺导向的芳基醚胺化方案. 在这里, Ti(NMe2)4既是金属催化剂, 又是氮的来源, 受到以往Ti(NMe2)4可作为C—H键活化剂的灵感[55], 结合苯酚衍生物通过芳基C—O键的断裂活化, 开发了此胺化方案. 反应普适性好, 供电子、缺电子取代基芳基醚均能以良好产率胺化, 除二甲氨基外, 二乙胺基和苯胺基也是此方案的良好氨源. 胺化过程的可能机制如Scheme 23所示. 吡咯甲基取代的亚胺[N-(2-甲氧基亚苄基)-1-(1H-吡咯-2-基)甲胺, 1]作为底物是芳基醚胺化成功的关键. 首先Ti(NMe2)4与1反应得到钛络合物I, 其金属中心采用伪八面体几何结构表示. C—O键活化过程中, 与甲氧基氧原子处于顺式的NMe2基团的氮原子可能非常接近带有甲氧基的碳原子, 此时C、N、O和Ti原子的相互作用产生四元环过渡态II, 随后经历C—O键断裂和C—N键形成过程, 生成络合物III, 最后水解得到终产物. 当甲氧基与亚胺导向基团处于间位时, 由于Ti—O和C—N距离大大增大, 因此, 预期的四元Ti−N−C−O环无法生成, 便无法完成C—O键活化, 说明了此反应的区域选择性.
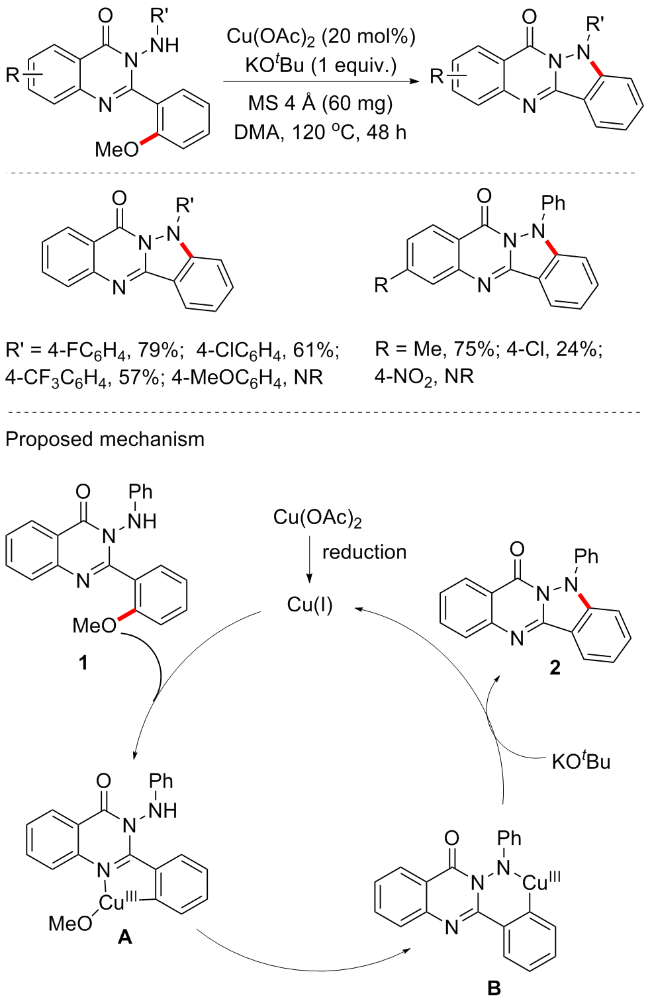
喹唑啉酮稠合杂环是一类重要的含氮杂环, 已被证明是一类重要的生物碱, 许多具有生物活性的产物都包括此结构, 如环糊精等. 2017年, 吴华悦等[56]实现了铜催化的芳基C—O键断裂形成分子内C—N键, 从而构建吲唑并[3,2-b]喹唑啉酮衍生物(Scheme 24). 新合成的吲唑并[3,2-b]喹唑啉酮的结构通过X射线晶体衍射分析得到了证实. 反应条件较为绿色环保, 无需配体, 但底物普适性和产率均一般(17例子, 产率24%~91%), 且含供电子取代基的芳胺不可耐受. 根据控制实验, 作者提出反应机制如下: 活性Cu(I)中间体首先通过亲核试剂还原Cu(II)或Cu(III)歧化而形成; 然后, 2-(2-甲氧基苯基)-3-(苯基氨基)喹唑啉酮(1)与Cu(I)盐的氧化加成将生成关键的C—O插入Cu(III)中间体A, 该中间体A可以随着温度的升高而经历“翻转”环金属化, 通过与H—N(Ph)喹唑啉酮的分子内配体交换提供六元中间体B, 最后C—N键形成提供吲唑并[3,2-b]喹唑啉酮, 并使Cu(I)催化剂再生完成循环.
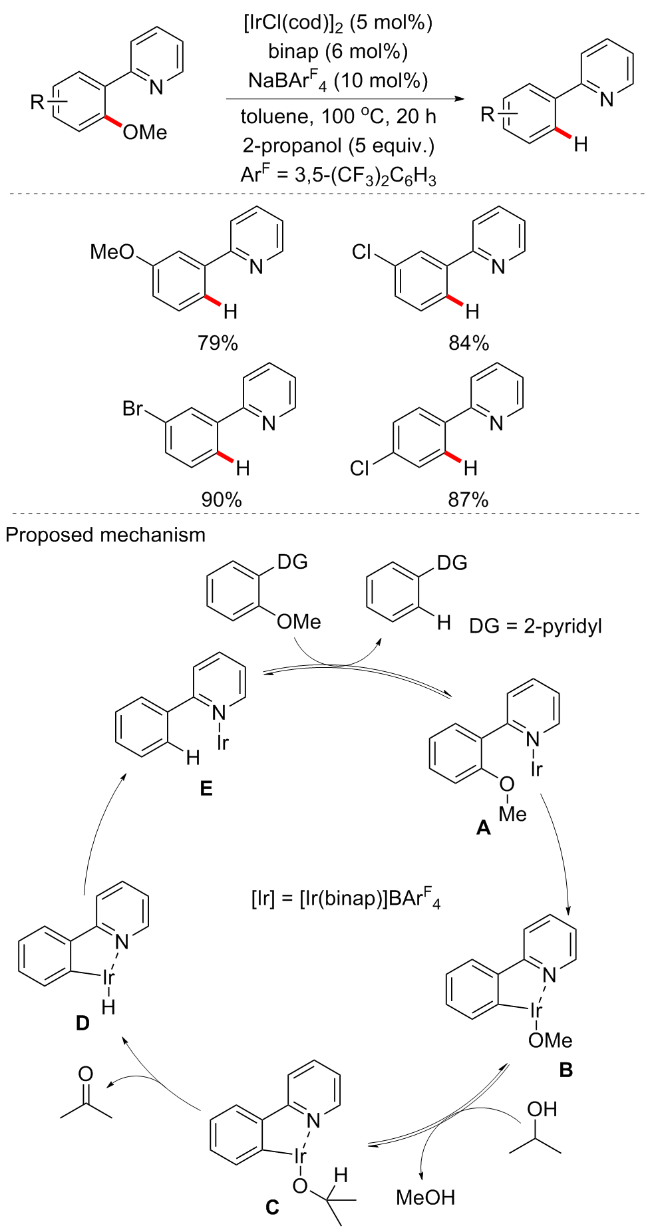
2-吡啶基团作为单齿导向基团在C—H键活化方面的应用已经较为成熟, 而金属铱经常作为光敏剂在光催化方面的应用较多. 2017年, Nishimura等[57]报道了金属铱(作为过渡金属催化剂)/磷催化剂(binap)可以在醇作为还原剂存在下催化以2-吡啶基作为导向基团的芳基醚的C—O键还原氢化方案(Scheme 25). 最初在[IrCl- (cod)]2 (5 mol%)、binap (6 mol%)和NaBArF4 (ArF=3,5- (CF3)2C6H3)存在下, 在100 ℃的甲苯中处理2-(2-甲氧基苯基)吡啶20 h, 得到产率为19%的2-苯基吡啶. 当添加5 equiv. 2-丙醇后, 反应产率显著提高, 以97%的产率得到目标产物. 环戊醇和乙醇作为还原剂也有效, 但没有异丙醇效果好. 此外, 用于形成阳离子铱的NaBArF4对于反应是必不可少的, binap是该反应最佳配体. 其他双膦配体, 如2,2'-双(二苯基膦酰基)-1,1'-联苯(dpbp)、4,5-双二苯基膦-9,9-二甲基氧杂蒽(xantphos)、1,1'-双(二苯基膦)二茂铁(dppf)和1,4-双(二苯膦基)苯(dppb), 以及单膦配体三苯基膦和三环己基膦在该反应中的有效性较低或无活性. 通过吡啶氮与阳离子Ir(I)配位, 并与邻位C—O键活化形成甲氧基铱(III)物种B, B上的烷氧基与2-丙醇交换生成异丙氧基铱C是该方案的关键步骤. 此外, 早期王键吉等[58]还报道了Fe催化芳基醚还原氢化方案.
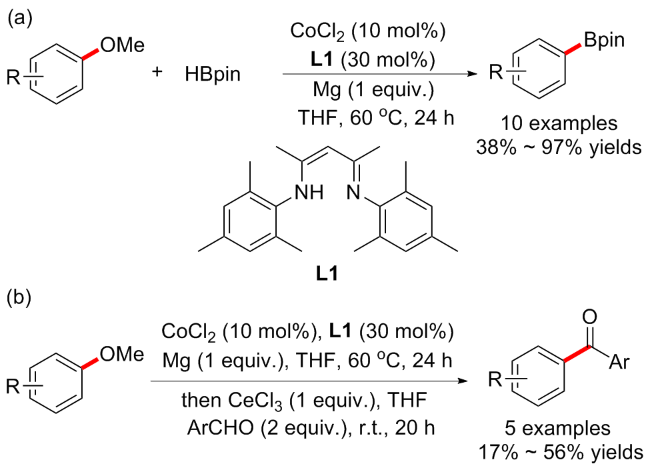
2022年, Lee等[59]报道了钴催化的C—OMe键官能化反应. 当在CoCl2 (10 mol%)、nacnac型配体L1 (30 mol%)和Mg (1 equiv.)为组合的催化下, 以HBpin (2 equiv.)为底物, 可实现芳基醚的硼化方案(10例子, 产率38%~97%) (Scheme 26a); 为了扩大此方案的转化范围, 作者在上述条件下反应后, 继续添加CeCl3作为催化剂, 苯甲醛作为底物, 选择性地提供相应的苯甲酰化产物(5例子, 产率17%~56%) (Scheme 26b). 力学研究表明, 格氏试剂是该反应的关键中间体, 通过C—O键活化反应生成, 电喷雾质谱法(ESI-MS)分析直接证实了格氏试剂中间体的存在. 有趣的是, 芳基硫醚在该体系下也能通过C—SMe锻炼, 以良好的产率转化为硼烷化物.
2 无过渡金属介导/催化的C—OMe键转化
2.1 自由基介导的C—OMe键转化
尽管过渡金属催化实现碳杂键的偶联在有机合成中一直具有举足轻重的地位, 但仍有改进的空间, 包括避免使用贵重金属(最终会产生金属废物)和昂贵的配体. 随着可持续发展化学的重要性日益显现, 无过渡金属催化方案似乎更符合当今绿色化学研究这一大主题.
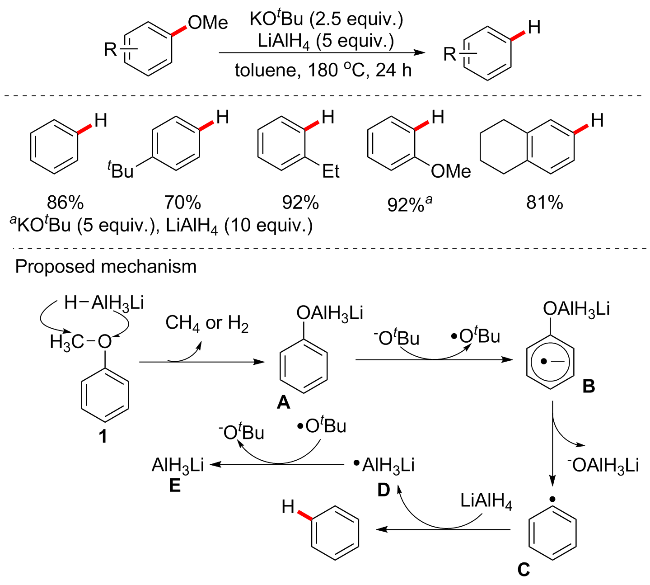
LiAlH4是一种强还原剂(或氢化物供体), 已广泛应用于有机合成中, 并显示出强大的还原能力[60]. KOtBu作为有机碱还可引发自由基并促进一些有机反应[61]. 2015年, 刘志敏等[62]发现LiAlH4和KOtBu的组合对活化包括酚类在内的各种芳香化合物中的芳基C—O键非常活跃, 而不需要使用任何其他催化剂或添加剂(Scheme 27). 在KOtBu (2.5 equiv.)存在下, 使用LiAlH4 (5 equiv.)作为还原剂, 可实现芳甲醚C—O键的选择性裂解以产生芳烃. 底物普适性良好, 各种供电子或烷烃取代基芳基醚均能在此条件下还原裂解, 此外, 苯酚等酚类衍生物也是此方案的良好底物之一, 而且具有可以将木质素化合物中常见的芳基C—O—C键解聚为芳烃的强大能力, 以良好至优异的产率获得所需的产物, 但不适用于含有缺电子取代基的底物, 并且当以2-萘甲醚为底物时, 芳环被氢化为环己烷. 考虑到KOtBu可引发自由基, 当添加TEMPO等试剂时, 反应被明显抑制. 基于上述实验结果, 作者推测反应可能是通过自由基机制进行, 并提出了LiAlH4-KOtBu体系催化芳醚脱氧的初步机制.
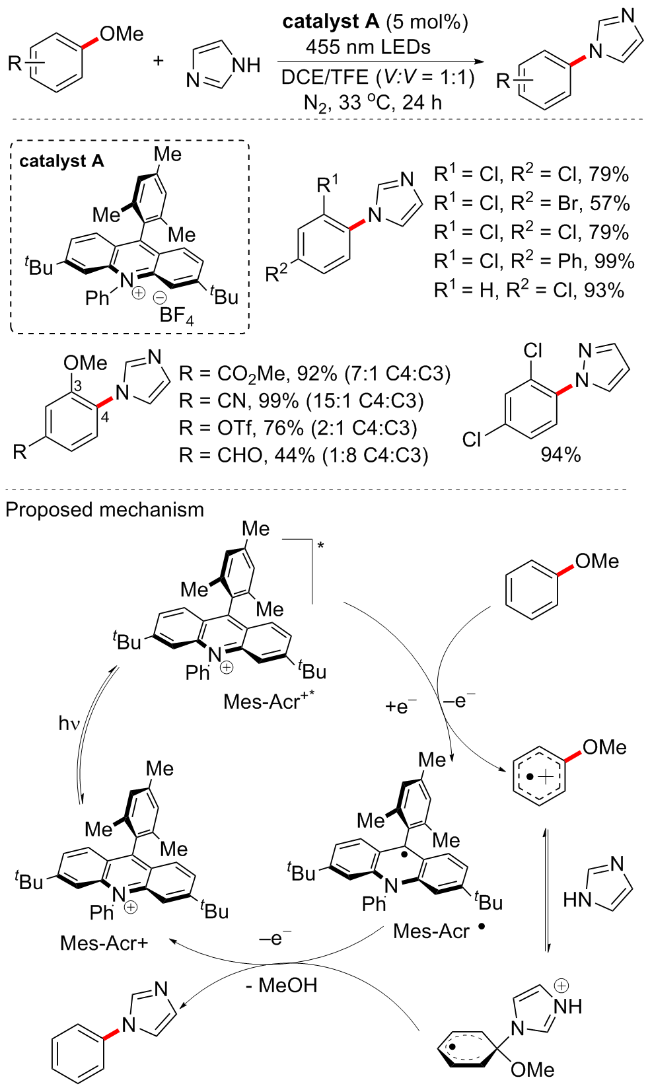
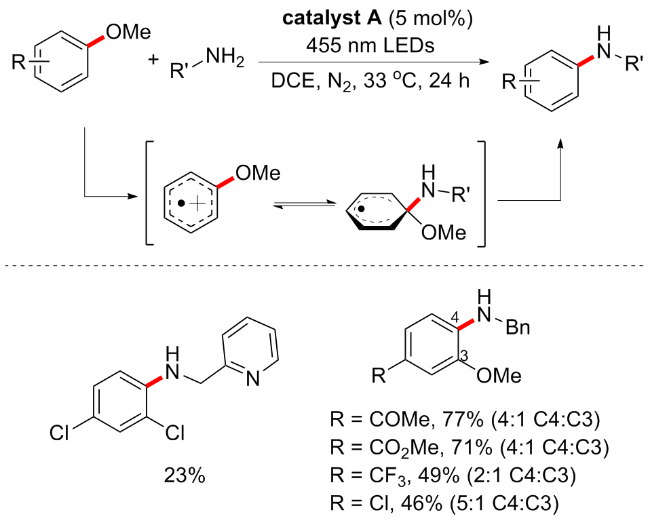
随后, Nicewicz等[63]开发了一种新型有机光催化剂催化的芳甲醚胺化方案, 是一种阳离子自由基加速的烷氧基芳烃的亲核芳香取代反应(Scheme 28). 因为具有高还原电势的3,6-二叔丁基-9-均三甲基苯基-10-苯基吖啶四氟硼酸盐(catalyst A, E1/2red*=+2.15V vs. SCE)可能是一个理想的催化剂, 激发态下可氧化得到关键的芳香烃阳离子自由基中间体. 作者首先以2,4-二氯苯甲醚为芳香底物, 咪唑为亲核试剂, 在吖啶光催化剂A条件下, 2,2,2-三氟乙醇(TFE)和1,2-二氯乙烷(DCE)的等体积混合物中使用过量的咪唑, 可以95%的产率形成所需的胺化芳烃. 对照实验确定了吖啶光催化剂A的重要性, 因为在没有光和光催化剂的情况下不会形成胺化产物. 反应底物普适性是良好的, 咪唑、吡唑和三氮唑等杂环胺都是该方案的良好底物, 缺电子取代基的芳基醚较供电子基表现更好, 但当3,4位同时存在OMe时, 在标准条件下产生一定比例的C4∶C3混合物. 最后, 根据相关实验, 作者提出的反应机制如下: 光催化剂在蓝光照射下跃迁激发态Mes-Acr+*, 此时具有很强的氧化性(E1/2red*=+2.15V vs. SCE), 可以将芳甲醚氧化成对应的自由基阳离子, 同时催化剂还原得到Mes-Acr•; 咪唑与芳甲醚自由基阳离子反应并脱去一分子甲醇得到胺化产物, 同时得到还原态的光催化剂A, 由此完成催化循环.
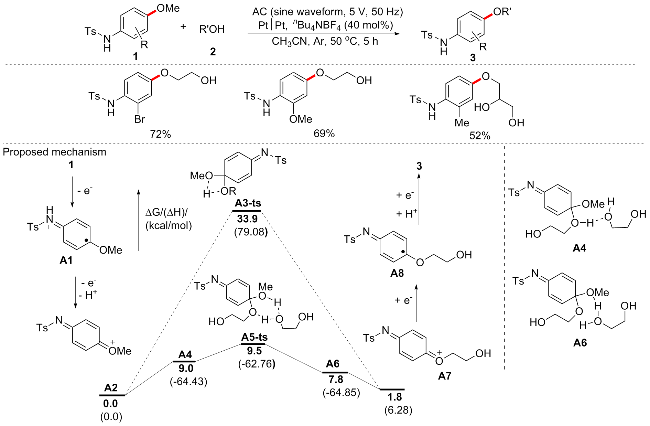
随着绿色化学研究的推进, 除光催化外, 电催化也成为近年热点之一, 因为有机电合成是建立绿色和可持续工艺的一种通用方法, 危险和有毒的氧化还原试剂被电流取代, 并且总体能耗降低. 2022年, 雷爱文等[66]报道了一种通过交流电解(AC)实现正式的芳甲醚C—OMe/醇O—H交叉复分解的新方案(Scheme 31). 该方案以温和的反应条件为特点, 允许容易获得的4-烷氧基苯胺和醇以高度区域选择性和化学选择性的方式转化为各种有价值的产物. 同时, 与以往只能有一个电极用于转化底物的直流电解(DCE)相比, 交流电解效率更好. 作者以N-甲苯磺酰基-2-甲基-4-甲氧基苯胺和乙二醇作为模板底物来研究AC促进的C—O/O—H复分解. 正如预期的那样, 当以nBu4NBF4为电解质, CH3CN为溶剂, 家用变压器(正弦波, 50 Hz)为电源进行交流电解(ACE)时, C—O/O—H交叉复分解反应顺利进行, 可以87%的产率产生所需的产物. 底物普适性广阔, 产率中等至良好, 但该反应大多限于对位有N-甲苯磺酰基等氮取代的底物, 而且邻甲氧基和间甲氧基底物不适用. 通过DFT计算, 作者推测反应机制可能如下: 在交流电解条件下, 底物1在电极表面通过两步单电子氧化依次氧化为A1, 然后氧化为A2; A2可能直接与醇2反应形成四元环过渡态A3-ts (ΔG≠=141.8 kJ/mol), 然后通过释放甲醇产生阳离子A7, 但A3-ts的活化自由能对50 ℃下的任何反应来说都太高, 并且能垒很可能是由四元环中的张力引起的. 因此, 作者继续考虑了质子转移路径. 首先, A2通过亲核加成和氢键与2的两个分子反应形成中间体A4; 随后, 发生分子内质子转移, 通过过渡态A5-ts (ΔG≠=39.7 kJ/mol)形成中间体A6; A6通过释放自由能降低25.1 kJ/mol的甲醇进一步转移到稳定物种A7. 尽管整个反应稍微吸热7.5 kJ/mol, 但在同一电极上通过极性反转立即还原A7, 使整个反应顺利进行, 从而提供最终产物.
2.2 Brønsted酸/碱催化的C—OMe键转化
2.2.1 Brønsted酸催化的C—OMe键转化
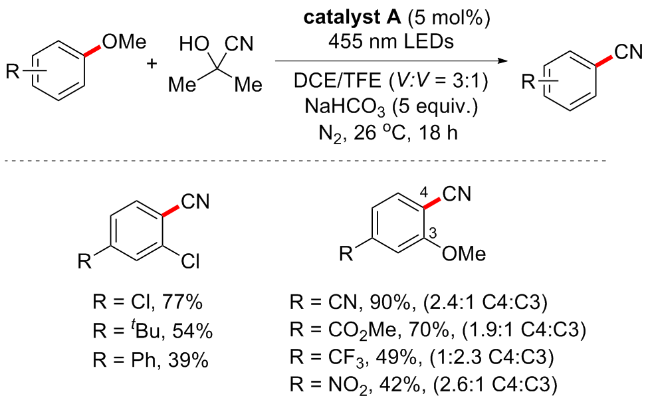
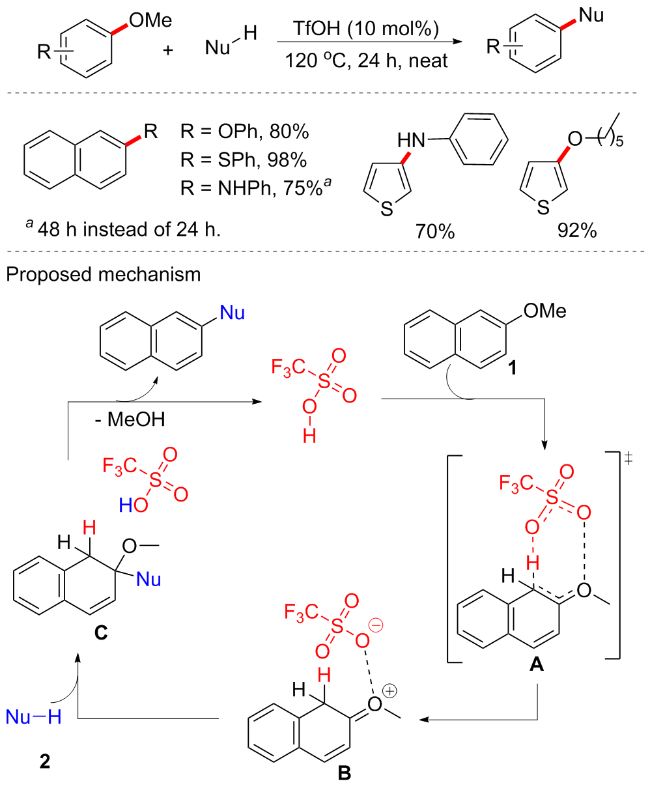
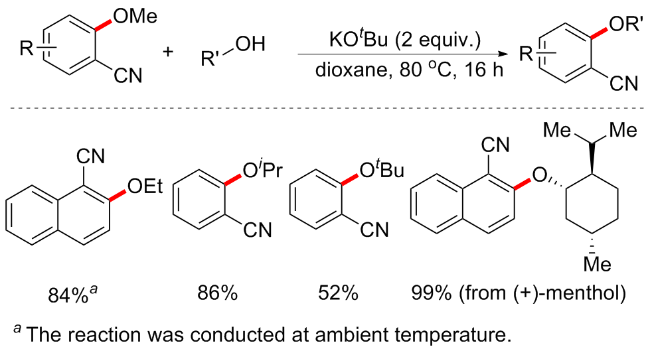
2017年, Biswas等[67]开发一种在无过渡金属催化条件下Brønsted酸催化的C—OMe键转化方案(Scheme 32). 该方案是在无金属、无配体、无溶剂条件下进行的高原子效率转化反应. 通过筛选不同的Brønsted酸, 如对甲苯磺酸(p-TSA)、甲磺酸(MeSO3H)、次膦酸(H3PO2)、乙酸(AcOH)、对氨基苯磺酸(H2NC6H4SO3H)、三氟乙酸(CF3CO2H)和三氟甲磺酸(TfOH), 最终发现10 mol% TfOH是该反应的最佳催化剂, 产率可达95%, p-TSA和H2NC6H4SO3H分别以52%和36%的中等收率实现芳甲醚的转化, 而其他Brønsted酸则不适用. 该方案底物兼容性佳, 包括O、N和S等中心的偶联试剂均可耐受, 并具有良好的收率(31例子, 产率52%~96%). 为了验证机理, 作者进行一系列氘标记实验, 如2-甲氧基萘在催化剂存在下与重水反应时, 仅在C1位置观察到67%的氘掺入; 类似地, 对于1-甲氧基萘仅在C2位置观察到氘掺入. 基于氘交换实验和以前相关研究, 推测机制可能如下: TfOH首先使1中的C1质子化, 质子化过程应该通过环状过渡态A发生; 亲核试剂对中间体B的C2中心进行亲核攻击, 随后消除甲醇, 在随后的反应步骤中通过催化剂的再生产生目标产物.
2.2.2 Brønsted碱催化的C—OMe键转化
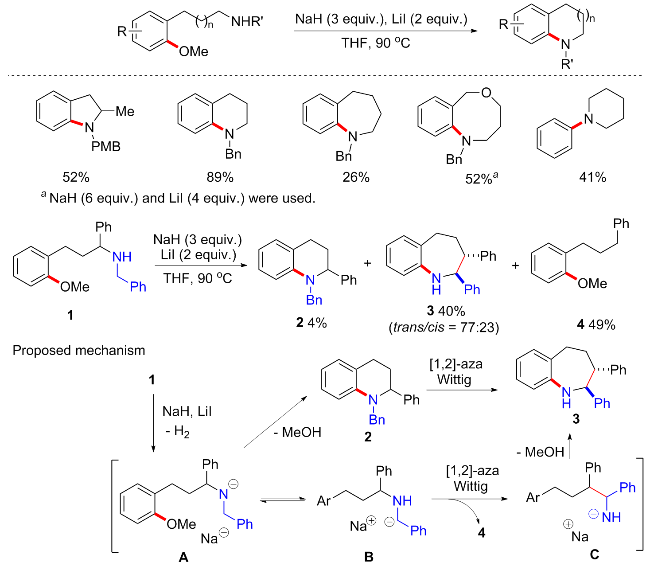
2017年, Chiba等[68]开发了一种Brønsted碱催化甲氧基芳烃的亲核胺化方法(Scheme 33). 反应最优条件为NaH (3 equiv.), LiI (2 equiv.)条件下, 以THF为溶剂, 90 ℃下反应4~47 h. 这种方法提供了一种制备苄环化氮杂环的有效途径, 适用于五元、六元、七元、八元甚至十元氮杂环的构建, 供电子和缺电子取代基的芳基醚也均可耐受. 此外, 对于吗啉、四氢吡咯和哌啶等也适用于分子间的胺化. 有趣的是, 当以带有苯基取代基的1为底物时, 可能是由于产生的阴离子电荷的离域作用, 目标产物仅4%, 会产生预期以外的产物3以及副产物4. 当缩小一个碳原子时, 还会逆Mannich反应产生副产物2-甲基苯甲醚和醛亚胺等. 作者推测含苯基取代的底物1首先经亲核芳香取代得到预期产物2, 随后可能进一步发生aza-[1,2]-Wittig重排, 通过苄基亚甲基部分的去质子化形成扩环产物3; 或者可以从苄基阴离子B中进行另一种aza-[1,2]-Wittig重排, 以形成新的C—C键生成氨基钠C, 最后氨基钠C环化得到扩环产物3.
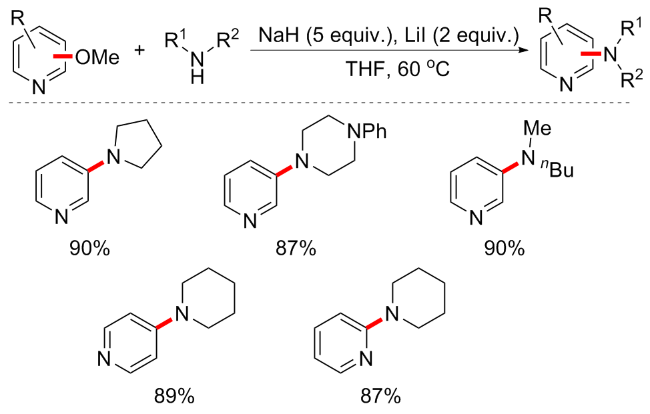
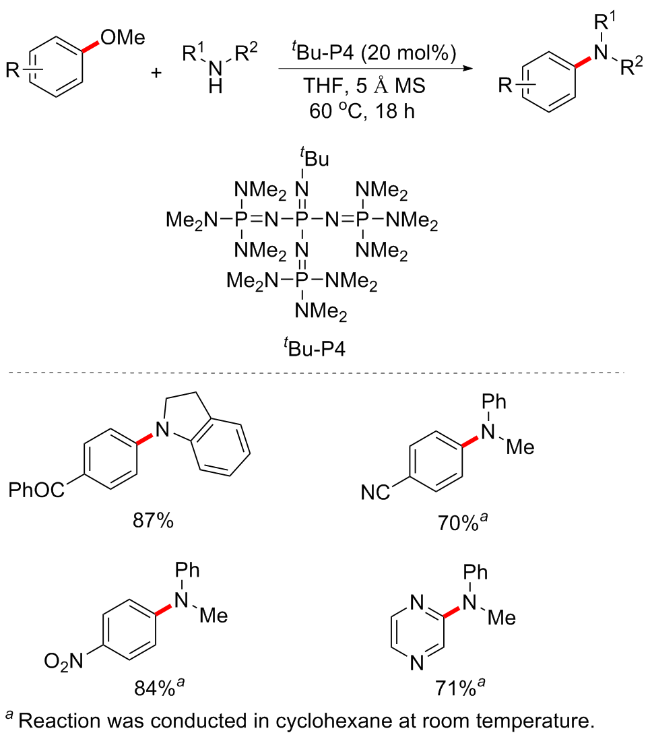
2019年, Kondo等[70]报道了一种有机超级碱tBu-P4 (Organic Superbase)催化的甲氧基芳烃与胺亲核试剂, 如苯胺、吲哚啉和氨基吡啶衍生物的胺化方案(Scheme 35). 有机超级碱的引入对于芳基醚的活化是非常有效的, 且条件绿色环保, 对于具有不同官能团(羰基、氰基、硝基和卤素)的缺电子甲氧基芳烃以及甲氧基杂芳烃(包括吡嗪、喹啉、异喹啉和吡啶衍生物)的转化均适用. 除分子间反应外, 分子内反应也可耐受, 并可合成六元环和七元环等环胺产物. 但对于供电子取代基的芳甲醚不适用. 同年, 该课题组还报道了有机超级碱催化的芳甲醚醇化方案, 并同样具有良好的底物普适性和收率[71].
2-芳基吲哚广泛存在于各类生物活性分子中, Snape等[73]曾对2-芳基吲哚发表了这样的评论: 在吲哚类化合物中, 2-芳基吲哚似乎是药物开发最有前途的先导. 最近, 毛建友和Walsh等[74]开发了一种碱促进的邻苯甲醚的苄基C—H去质子化, 将所得阴离子添加到苄腈中, 从而合成2-芳基吲哚(Scheme 37). 在不添加过渡金属催化剂的情况下, 可以良好的产率(>30例子, up to 99% yield)制备了多种2-芳基吲哚. 该方案对供电子取代基腈类表现较好, 但不适用于缺电子类苯甲腈(如卤素、三氟甲基等). 由于在二(三甲基硅基)氨基碱[MN(SiMe3)2]促进的反应中, MN(SiMe3)2既可以是去质子化试剂, 又可以作为氮源引入反应中, 我们课题组和Walsh等[75]都已证实. 在该报道中, 作者还通过在标准条件下使用15N标记的2-萘腈, 探索了吲哚产物中氮原子的来源. 基于吲哚产物的N—H (14N为单线态, 15N为双峰态)的1H NMR分析, 发现标记的15N与LiN(SiMe3)2的14N确实发生了交换. 来自腈的吲哚中的氮与LiN(SiMe3)2的比例约为1∶2. 结果表明, 交换作用比SNAr更快. 基于上述结果, 可能的反应途径如Scheme 37所示.
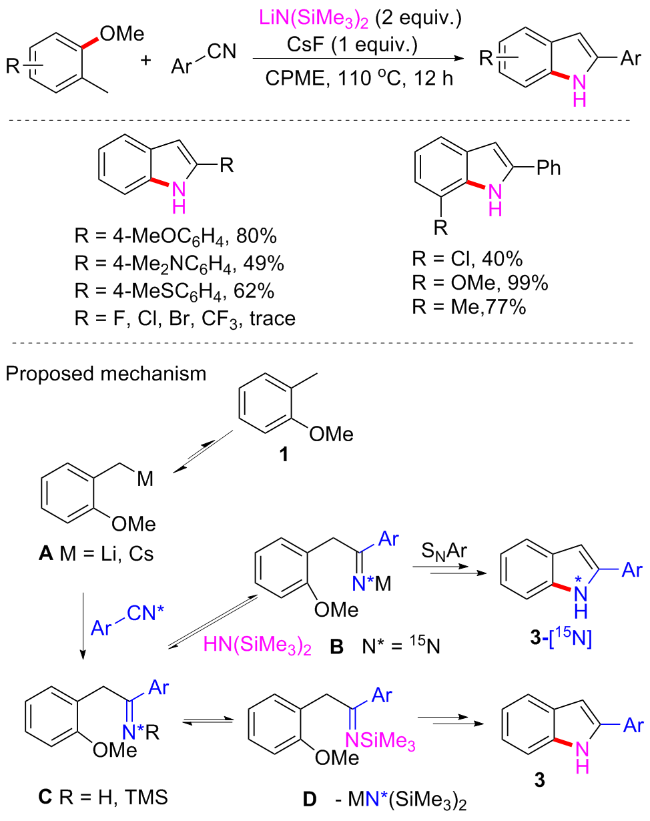
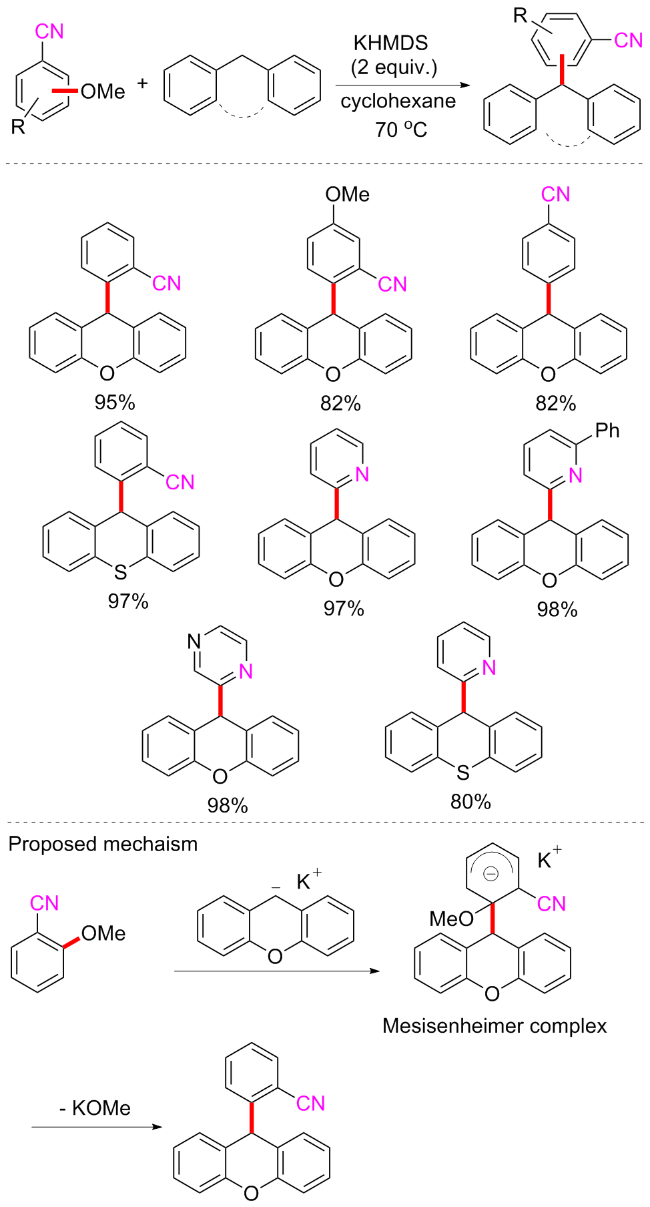
2023年, 赵万祥等[76]实现了Brønsted碱促进的二芳基甲烷和芳基醚的交叉偶联反应(Scheme 38). 该方案以氰基或者吡啶氮为定位基团, 在氰基或者吡啶氮邻位的C—OMe可与芳基甲烷偶联, 产率和普适性均非常优异, 且选择性良好. 二芳基甲烷类亲碳核试剂, 如9H-硫氧蒽、10-甲基-9、10-二氢吖啶、9H-芴和二苯基甲烷等均可在该方案中耐受, 而含有氰基的产物很容易进一步转化为羧酸、酰胺、酮和醛, 证明了该方案的实用性. 可能的反应机制为9H-氧杂蒽首先在双(三甲基硅烷基)氨基钾(KHMDS)下去质子产生亲核物质, 然后进攻2-甲氧基苯甲腈形成一个Meisenheimer复合物中间体, 最后经过甲醇钾离去得到目标产物.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 总结与展望
总之, 开发高效、绿色和简单的芳基醚交叉偶联反应是当前研究的热点与重点之一. 尽管目前已有大多数文献报道过芳基磺酸盐或芳基酯衍生物的C—O裂解偶联, 但从原子和步骤经济的角度来看, 在这些反应中产生大量废物和需要对起始苯酚进行衍生化是需要克服的重要缺点. 显然, 利用芳甲醚作为偶联试剂是向前迈出的一大步, 考虑到芳基醚是有机合成中普遍存在且用途广泛的基序, 也是芳基卤化物良好的替代品, 本文中展现的许多转化可以为后续合成提供使用芳基醚设计合成策略的新思路. 这些先前的研究结果也极大地丰富了有机合成方法, 为药物化学和药物的发展做出了巨大的贡献. 本文详细总结了近十年来基于C—OMe键断裂的芳甲醚转化反应研究进展, 根据不同催化条件和反应原理进行分类: 包括过渡金属催化的基于C—OMe键转化和无过渡金属催化/介导的C—OMe键转化(自由基介导的C—OMe键转化和Brønsted酸/碱催化的C—OMe键转化), 并详细论述了反应条件、底物耐受性、反应机理、应用与不足等.
尽管最近取得了显著的进展, 但基于芳甲醚C—OMe键断裂的转化反应仍存在部分局限性: (1)昂贵金属(如镍)和配体的使用仍然占主导地位, 而廉价或不含过渡金属方案的使用频率较低, 并且仍处于初级阶段. (2)此外, 尽管目前金属镍催化剂在芳基亲电试剂的C—OMe键活化方面表现出优异的催化活性, 但最近的研究已经证明其他过渡金属如Cr、Fe、Co、Ru和Cu催化C—OMe键断裂的潜力. 对这些金属在C—OMe活化中的反应性和行为进行系统研究, 将有助于更好地理解C—OMe氧化加成机制, 并开辟更新型的C—OMe活化模型. (3)尽管过渡金属至关重要, 但配体通常也是控制反应性的关键, 最近的研究表明, NHC配体(如IMes、IPr、I(2-Ad)等)对促进C—OMe活化很重要, 因此开发更多类型的NHC配体, 搭配过渡金属催化将有可能进一步提高芳基醚转化效率. (4)反应底物通常存在一定局限性, 一类反应或者仅限于供电子取代基芳基醚, 或者仅限于缺电子取代基芳基醚, 此外这些方法在实际合成中的应用仍然非常理想. 如今, 高效率催化、生物催化和流动催化等可以有效地提高有机合成反应的速率和效率, 并能提高产物的纯度和产率, 减少副反应和废物的产生, 这类方法有望成为此类反应的突破方向. 总之, 以芳甲醚为芳基化源, 在温和的反应条件下, 以更经济、更环保的方式实现交叉偶联的可持续技术的发展仍有广大的前景.
(Lu, Y.)