


胺类化合物广泛存在于天然产物、生物活性分子以及相关药物分子中[1]. 在美国亚利桑那大学Njarðarson研究团队发布的2021年全球小分子药物销售额前200排行榜中, 接近一半药物分子同时含有胺基结构和手性中心[2]. 碳氢键活化策略的发展为这些分子的合成和修饰提供了便捷途径. 其中, 发展可调控的区域和对映选择性碳氢键官能团化新体系, 高效绿色地在胺类化合物不同位置(α, β, γ等位置)直接引入官能团, 已经成为有机合成中的一个研究重点, 但仍然面临许多挑战[3].
首当其冲需要解决的是不同位点C—H键的识别以及反应活性的问题. 目前, 选择性一般在氮原子的辅助或影响下进行控制, 同时需要引入可离去的辅助或保护基团(一般含酰胺结构). 化学家们早期发展了杂原子导向策略, 利用安装导向基团或者利用底物固有胺基导向基团配位金属催化剂, 然后分子内活化目标C—H键, 通过形成不同尺寸的环金属中间体, 实现不同位置选择性的官能团化反应[4]. 该策略通常体现出优异的区域选择性, 但一般需要在相对剧烈的条件下进行, 且反应类型相对局限[5]. 与之相比, 基于氢原子转移(HAT)的自由基攫氢策略可以在温和条件下实现目标C—H键的氢转移和官能团化过程, 因此在近几年来受到了广泛关注(Scheme 1, a)[6].
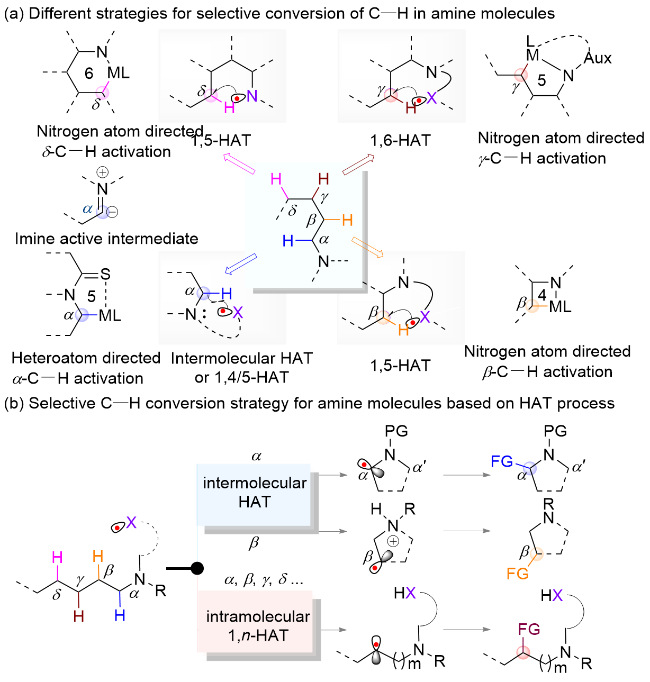
HAT是指在化学反应中氢原子从一个位置转移到另一个位置的过程, 其本质上涉及自由基物种的转换, 一般为高活性自由基物种攫取化合物C—H键生成碳自由基[7]. 通常, 胺类分子的HAT可以分为分子间过程和分子内过程. 其中, 受氮原子的活化影响, 胺类的分子间HAT主要发生在α位, 而通过安装不同类型的辅助基团则可以经分子内1,n-HAT过程实现胺类不同位点包括α位的选择性氢转移官能团化(Scheme 1, b)[8]. 近些年来, 随着对HAT过程机理的深入理解以及新型HAT试剂的开发, 基于HAT过程的胺类化合物选择性C—H键转化反应取得了显著进展, 因此亟需对相关反应体系进行及时总结和分析. 目前, 现有关于胺类化合物的自由基反应综述多聚焦于分子内HAT介导的化合物远端官能团化反应, 而对近端C—H键转化、分子间HAT过程及其选择性控制策略等方面尚缺乏全面综述[6a,6c].本文将根据胺类化合物不同位点详细综述近十年来报道的典型案例, 并针对其中的区域选择性、化学选择性和立体选择性控制策略以及反应机制进行总结和评述, 分析其存在的问题和挑战, 并对未来研究方向进行展望.
1 氮α位C—H键官能团化
由于氮原子的超共轭作用和电性影响使胺类化合物α位C—H键具有较低的键解离能(BDE, ≈384.6 kJ/mol), 因此易于发生攫氢过程, 同时形成稳定的氮α-碳自由基[9]. 因此, HAT介导的氮α位官能团化反应发展相对较多, 且具有条件温和、选择性好及转化多样等优点, 成为了有机合成领域的研究热点之一.
1.1 分子内攫氢
1.1.1 碳氮键引发
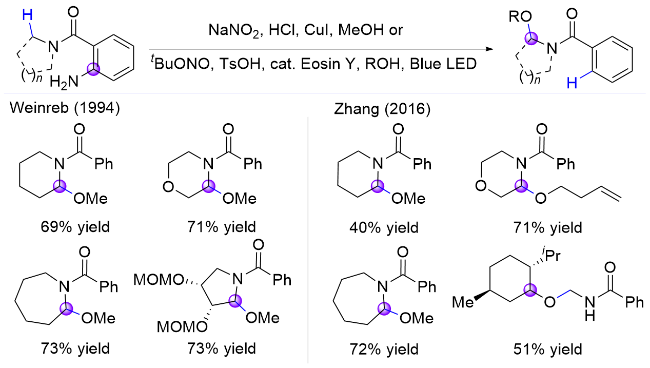
1.1.2 碳碘键引发
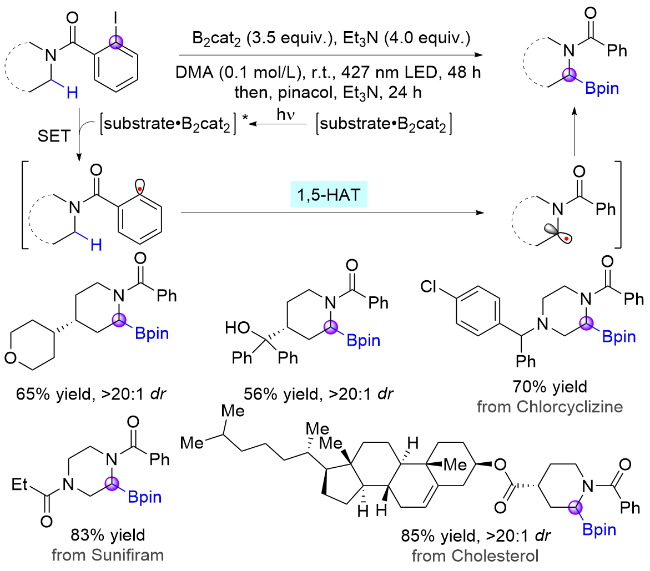
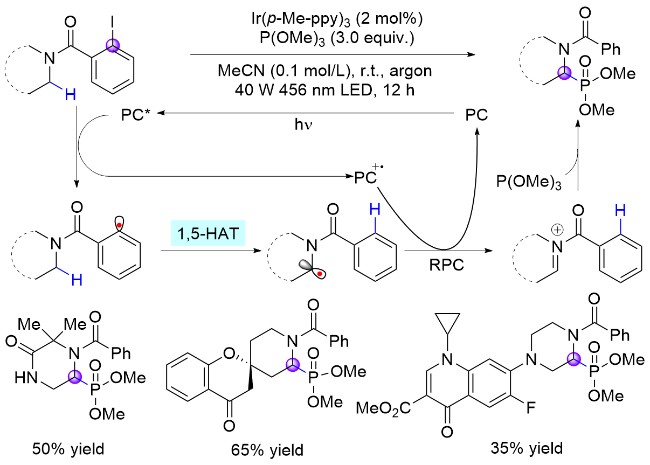
通过重氮盐产生芳基自由基通常需要比较复杂的前置操作, 同时需要氧化剂和酸性条件, 不符合绿色化学的理念. 近年来新兴的电子给体-受体(EDA)复合物提供了另一种解决该问题的新视角: 无需依赖光催化剂, 由电子供体和受体通过非共价相互作用形成的EDA复合物在光照下跃迁至激发态并发生电子转移, 即可产生目标自由基中间体, 继而完成相应转化[12]. 2022年, Gevorgyan课题组[13]使用碘代苯作为芳基自由基前体, 利用苯甲酰胺底物与儿茶酚硼烷络合形成的EDA复合物可在光诱导下发生C(sp2)—I键均裂, 实现非金属催化的脂肪胺α-C—H键选择性硼化反应(Scheme 3). 该策略不仅具有反应条件温和、底物范围广、官能团耐受性好以及区域和非对映选择性高等优点, 还能实现生物活性分子的后期硼化. 基于类似机制, Wu课题组[14]使用铱催化剂代替硼烷, 在光照下攫取碘原子生成苯基自由基, 然后经历1,5-HAT生成氮α-碳自由基以及自由基-极性交叉(RPC)过程, 实现了苯甲酰基保护的氮α位磷酸化反应, 突破了之前体系只适用于硼基化反应的限制(Scheme 4).
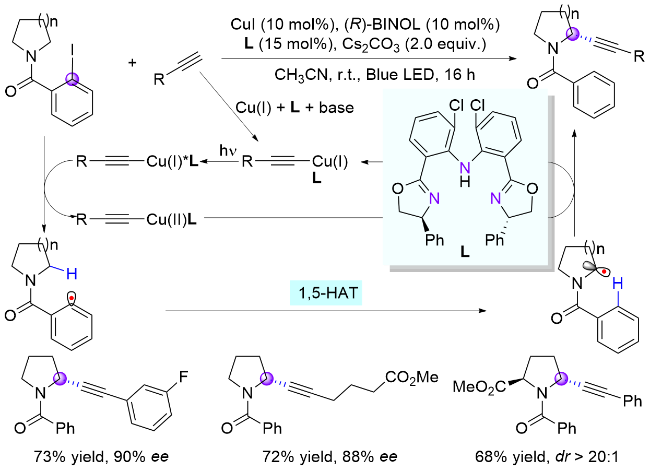
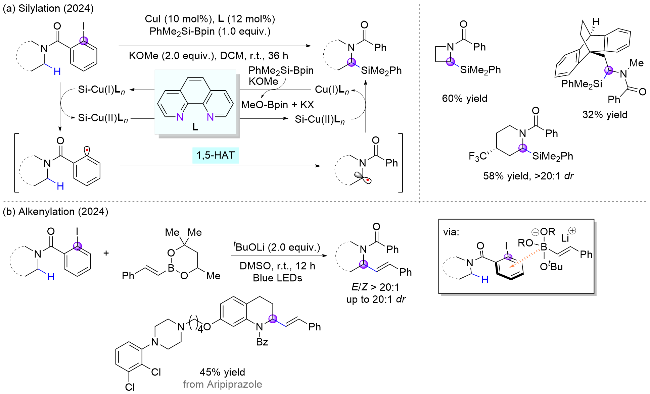
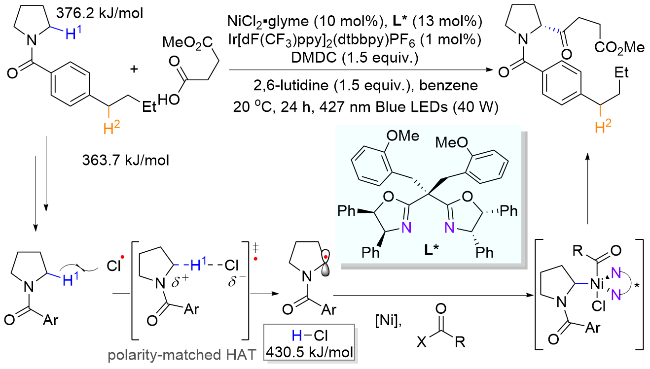
2024年, 郭瑞课题组[17]报道了两例苯基自由基介导的氮α位官能团化反应, 分别成功引入了硅基和烯基官能团. 其中, 在第一例反应使用廉价易得的碘化亚铜作为氧化还原催化剂, 在无光照条件下进行单电子氧化生成苯基自由基(Scheme 6, a). 而第二例反应借助了硼烷在碱参与下与底物形成具有光学活性EDA复合物的性质, 开创性地以烯基硼酸酯作为电子给体, 2-碘苯甲酰胺为电子受体, 在无需过渡金属和光催化剂的条件下实现了光诱导EDA复合物促进的氮α位C(sp3)—H与烯基硼酸酯的交叉偶联反应, 构建了一系列烯丙基胺化合物(Scheme 6, b)[18].
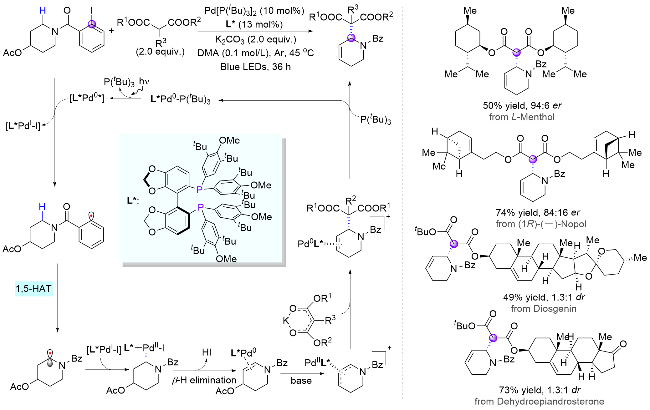
过渡金属催化的不对称烯丙基化取代反应在构建碳-碳键和碳-杂原子键中应用广泛, 是重要的有机合成方法之一[19]. 然而, 此类转化一般需要使用预官能团化的烯丙基结构单元. 2025年, 阳华课题组[20]巧妙地结合HAT介导的光催化远端C(sp3)—C(sp3)键去饱和过程与不对称烯丙基取代, 实现了哌啶衍生物α位对映选择性烷基化构建手性C—C键(Scheme 7). 该反应通过光激发钯物种[LPd0]对碘苯C—I键的单电子转移完成苯基自由基的生成, 然后分子内迁移后得到的氮α-碳自由基再与一价钯[LPdI-I]结合, 并经β-氢消除和醋酸根的离去原位生成烯丙基钯物种, 最后在手性配体辅助下发生不对称亲核取代反应得到目标产物. 该反应具有底物范围广泛、收率良好(最高96%收率)、α位选择性优异和对映选择性出色(最高91% ee)等特点.
1.1.3 碳溴键引发
一般而言, 分子内HAT遵循过渡态能量最低原则. 由于1,5-HAT所形成的六元环椅式构象过渡态具有较低焓势垒(enthalpic barrier)和熵势垒(entropic barrier), 所以其反应速率最快[21]. 因此, 该分子内氢迁移策略多用于实现δ (C-5)位的官能团化. 不过, 当伯碳氢键和苄位碳氢键同时存在时, 具有更低解离能的苄基碳氢键更容易发生攫氢过程(>418 vs 376.2 kJ/mol), 反应模式也不再局限于1,5-HAT. 借助该性质, Nishikata课题组[22]使用不含C-5氢的底物, 同时通过调整配体空间位阻及电子效应, 克服分子内1,4-HAT的高焓势垒, 实现了叔碳自由基介导的苄胺衍生物α位醚化反应(Scheme 8).
1.1.4 碳金属键引发
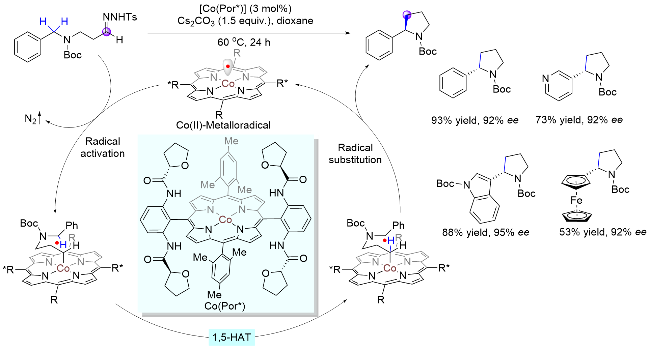
三线态金属卡宾由于每一个p轨道只有一个电子, 从而具有自由基的性质. 因此, 卡宾类金属碳自由基也可以经历分子内1,5-HAT生成氮α-碳自由基. Zhang课题组[23]在2018年提出了一种Co(II)金属自由基催化(MRC)的策略, 用于分子内对映选择性环化构建手性五元环(Scheme 9). 在该体系中, 首先手性卟啉钴插入底物的重氮基团, 脱去一分子氮气后生成α-Co(III)碳自由基, 接着发生分子内自由基攫氢过程, 即C-5位氮α-碳上氢原子迁移至钴α-碳原子上, 从而形成氮α-碳自由基. 最后, 该自由基与钴α-碳原子发生对映选择性的环外叔碳环化反应, 构建一系列手性四氢吡咯类化合物. 该体系对各种官能团和杂芳基底物有较好的兼容性.
1.1.5 氮氧键引发
杂原子中心自由基介导的分子内1,2-HAT为α位 C—H键官能团化提供了另外一种的策略. 不过, 相对于常见的1,5-HAT反应, 1,2-HAT过程由于高能垒和三元环过渡态的约束较为罕见, 现有的例子一般多发生于氧自由基中心[8c,24]. 2023年, 羊晓东课题组[25]开发了一种碱辅助的氮自由基分子内1,2-HAT策略, 实现了酰胺氮α位C(sp3)—H键与氮杂烯丙基化合物的偶联反应, 在温和条件下高选择性地合成了一系列1,2-二胺类衍生物(Scheme 10). 作者通过一系列控制实验, 证明反应经历了自由基历程, 并且体系中同时存在氮α-碳自由基和氮杂烯丙基自由基. 另外, 当底物同时具有α、δ和ε- C—H键时, 反应仍然高选择性地发生1,2-HAT过程, 生成α位偶联产物. 密度泛函理论(DFT)计算表明, 酰胺氮自由基α-碳原子去质子化过程的过渡态能垒为6.3 kJ/mol, 显著低于直接的1,2、1,5或1,6-HAT过程的能垒(分别为120.8、20.1、221.5 kJ/mol), 说明氮自由基到氮α-碳自由基的转变机制为碱辅助的去质子再质子过程.
1.2 分子间攫氢
上述策略采用了分子内原位生成的自由基作为HAT试剂, 因此一般需要引入辅助基团使之满足分子内优势的HAT过程, 导致底物适用类型受限. 为了突破该限制, 研究人员设计开发了各种各样的自由基攫氢试剂, 以分子间HAT的方式生成氮α-碳自由基, 从而扩大了胺类化合物的适用范围.
1.2.1 含氮自由基介导
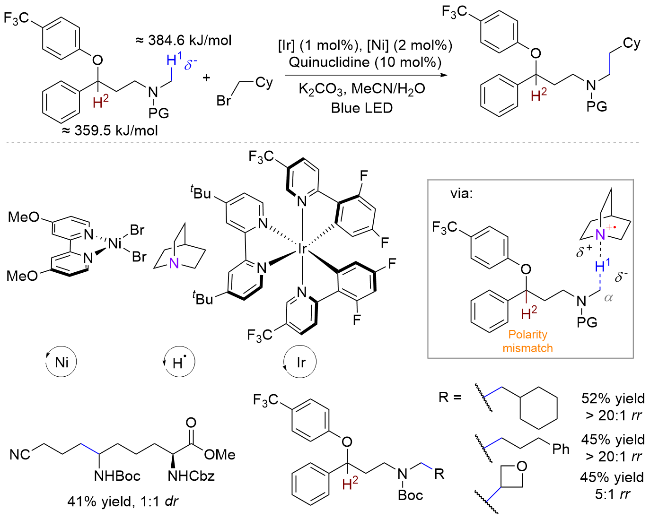
以光氧化还原的方式激发HAT试剂实现分子间选择性攫氢和自由基转化, 是一种理想的胺类化合物转化策略[27]. 2017年, MacMillan课题组[9]提出了极性匹配原理, 利用氮α位C—H键具有一定富电性的特点, 实现了其高选择性烷基化反应(Scheme 12). 在之前的认知中, C—H键均裂生成碳自由基的能力由其解离能决定, 因此HAT试剂攫氢的选择性一般跟解离能高度相关, 即数值越小, 越容易生成自由基. 然而研究发现, 虽然苄基C—H键具有更低的解离能(≈359.5 kJ/mol<≈384.6 kJ/mol)[28], 其烷基化却发生在氮α位. 作者认为, 在氮原子影响下α-C上氢原子呈现富电子性质, 与光激发奎宁环产生的缺电子氮自由基阳离子极性匹配, 因而优先攫取该位置的氢原子.
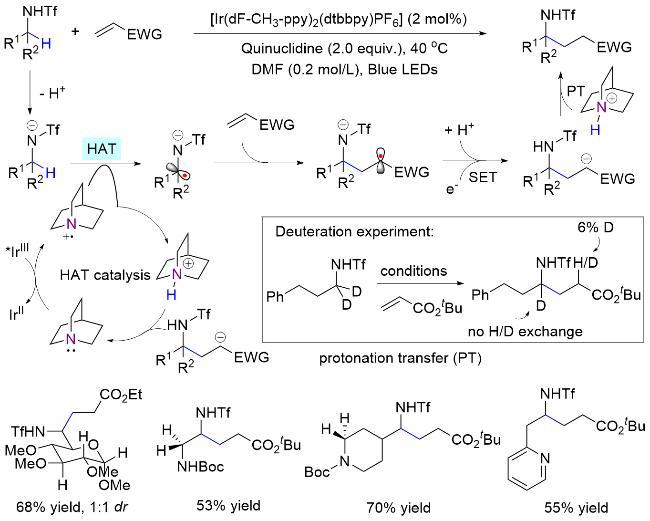
1.2.2 含氧自由基介导
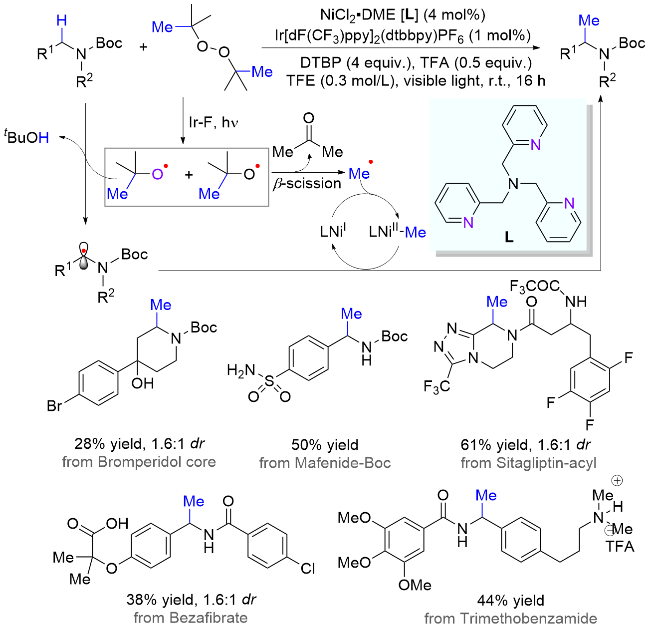
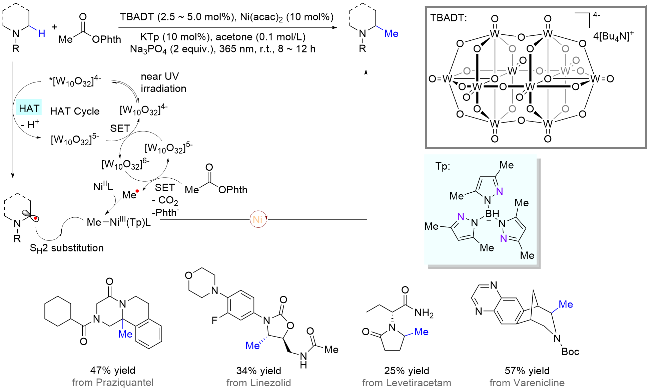
在药物研发工作中, 活性分子结构的微小改变有可能对其药理药效带来巨大的影响. 其中, 许多例子表明, 当药物分子中一个氢原子被甲基取代后, 很可能会提高其活性和稳定性, 同时降低毒性, 因此被称为“神奇的甲基效应(magic methyl effect)”. 所以, 基于快速修饰和筛选复杂分子结构, 提高研发效率, 开发兼容生物活性分子的直接后期甲基化方法显得尤为重要. 2021年, Stahl课题组[30]报道了一个光/镍协同催化利用过氧化物实现复杂分子的甲基化反应(Scheme 14). 该研究的设计思路是, 在三线态光敏剂的作用下, 过氧化物经能量转移断裂O—O键, 生成两分子的烷氧基自由基; 然后, 一分子的烷氧基自由基作为HAT试剂攫取氮α位C—H键, 而另一分子则裂解为甲基自由基并释放出丙酮分子; 最后, 镍催化剂可捕捉瞬态甲基自由基生成稳定的有机金属中间体, 从而顺利地与氮α-碳自由基发生选择性偶联反应. 气体成分分析表明, 当无镍催化剂时, 气体产物主要为甲烷, 说明此时甲基自由基主要参与HAT过程而不是自由基偶联过程; 而当镍催化剂存在时, 其气体主产物为乙烷, 证明反应过程中生成了甲基镍活性物种. 值得一提的是, 在该体系中许多复杂含氮药物分子顺利引入甲基官能团, 包括未修饰的伯胺和叔胺分子.
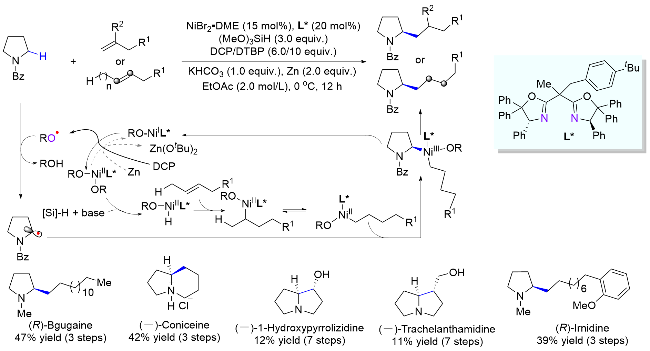
尽管通过分子间HAT/镍协同催化策略能实现胺类化合物的α位与亲电试剂的偶联反应, 其对映选择性烷基化构建手性C(sp3)—C(sp3)键仍然具有挑战性. 最近, 孔望清课题组[32]使用丰富易得的饱和氮杂环和烯烃为偶联试剂, 过氧化物为HAT试剂兼氧化剂, 金属锌为还原剂, 同时巧妙地借助Ni-H与烯烃的氢烷基化过程, 实现了一系列重要的手性含氮杂环的合成(Scheme 16). 其反应机理如下所示: 过氧化物氧化一价镍物种[RO-NiIL*]时分别生成了二价镍物种[(RO)2-NiIIL*]和一分子烷氧基自由基RO•, 而该自由基作为HAT试剂可攫取目标C—H键生成氮α-碳自由基并参与催化循环. 作者推测, 金属锌的作用可能是重新还原二价镍物种[(RO)2-NiIIL*]回到一价镍物种[RO-NiIL*], 然后继续被过氧化物氧化. 两者的循环转化可以增加溶液中自由基RO•的浓度, 从而提高HAT过程的效率. 该体系兼容各种未活化烯烃, 包括端烯、内烯和烯基硼酸酯. 值得一提的是, 该方法不仅可用于许多天然产物和药物分子的后期修饰, 也可用于合成一系列含氮杂环类生物活性分子.
1.2.3 含硫自由基介导
胺类化合物依据烷基取代基的数量分为伯胺、仲胺和叔胺, 其中仲胺与叔胺由于不同侧链的α位C—H键性质极为相似, 其不同取代基的选择性控制极具挑战, 因此之前的研究多着重于发展两边对称的环胺类底物的官能团化. 不过, 对于N-甲基-N-烷基类仲胺底物, 由于甲基位的位阻相对亚甲基更小, 且同时拥有三个C—H键, 更有机会发生攫氢过程, 因此自由基介导的α-胺甲基位官能团化产物往往占据主导(Scheme 17).
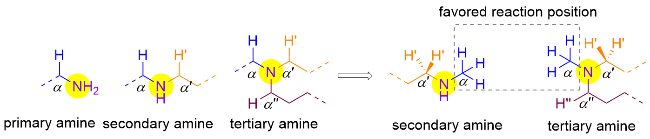
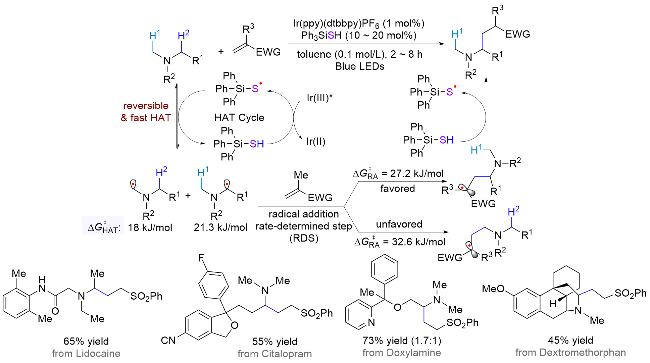
硫醇是一类极为有效的HAT催化试剂, 结合光催化体系生成硫自由基, 在催化循环中可高效攫取或提供氢原子[33]. 2021年, Rovis课题组[34]使用可逆的HAT催化机制, 利用硅硫醇强大的攫氢能力降低HAT过程的能垒, 从而逆转了选择性, 实现了叔胺化合物的亚甲基α位烷基化(Scheme 18). 作者通过密度泛函理论计算(DFT)发现, 不管是否加入催化量的硅硫醇, 甲基比亚甲基更倾向于发生HAT过程(甲基: 68.6或18.0 kJ/mol, 亚甲基: 73.6或21.3 kJ/mol). 然而, 与一般反应中HAT是决速步骤且过程不可逆不同的是, 体系中硅硫醇的加入显著降低了HAT过程的能垒, 使后续的自由基加成成为了决速步骤(RDS, 甲基能垒为32.6 kJ/mol, 亚甲基为27.2 kJ/mol), 也即产物决定步骤, 此时亚甲基位的烷基化能垒更低, 因此优先生成. 作者也通过氘代实验证明了HAT是可逆过程. 基于此机制, 该体系高收率地实现了一系列药物分子的后期选择性烷基化(45%~82%收率), 展现出了潜在的应用前景.
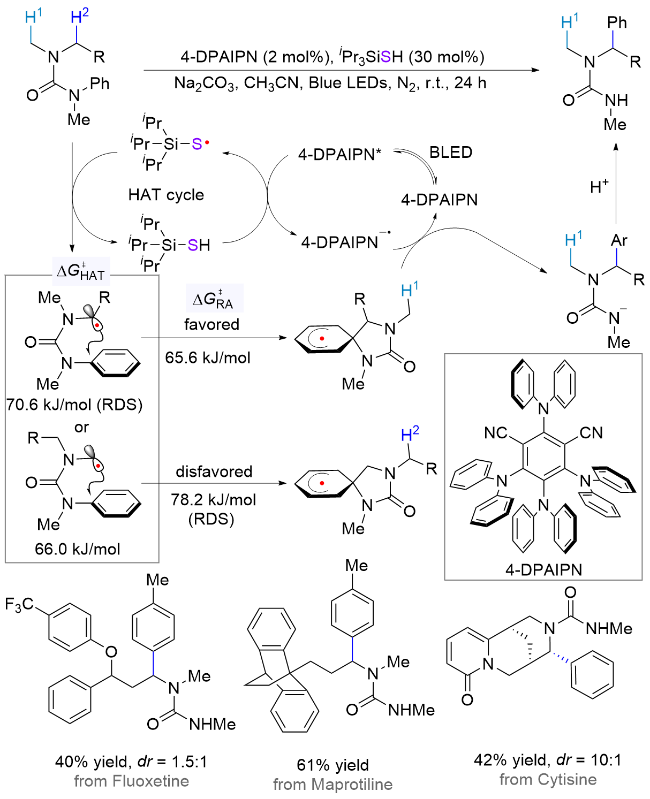
类似地, 蒋恒课题组[35]也通过控制反应的决速步骤实现了仲胺衍生物分子内芳基的α位选择性迁移反应(Scheme 19). 同样使用硅硫醇作为HAT试剂, 作者基于DFT计算证明, 虽然甲基位的攫氢过程能垒更低(66.0 kJ/mol vs 70.6 kJ/mol), 但是芳基迁移至甲基位需要越过78.2 kJ/mol的能垒(决速步骤), 而迁移至亚甲基位的能垒为65.6 kJ/mol, 显著降低. 对比整个催化循环的能量变化, 迁移至甲基位的能垒(78.2 kJ/mol)高于亚甲基位的攫氢能垒(65.6 kJ/mol), 因此促使反应发生在能量更低的亚甲基位. 该反应通过控制氢原子攫取和自由基加成两个基元步骤的能垒变化, 最终实现了胺基不同侧链α位官能团化选择性的逆转. 另外, 该体系也适用于许多仲胺药物分子的后期官能团化, 如氟西汀(抗抑郁,40%收率)、马普替林(抗抑郁, 61%收率)和金雀花碱(急救药, 42%收率), 同时具有优异的位置选择性.
1.2.4 卤素自由基介导
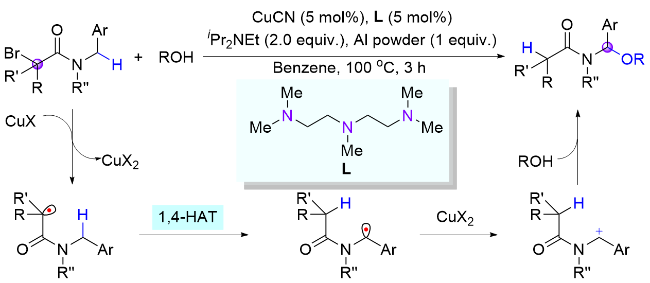
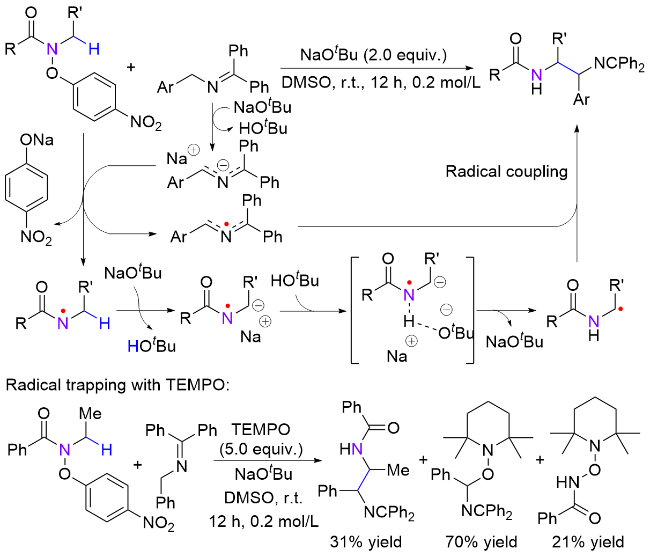
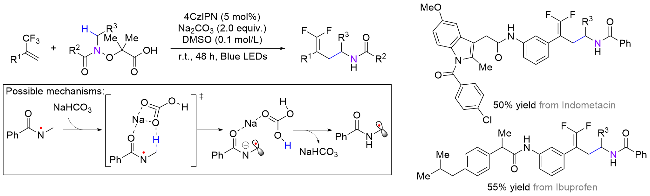
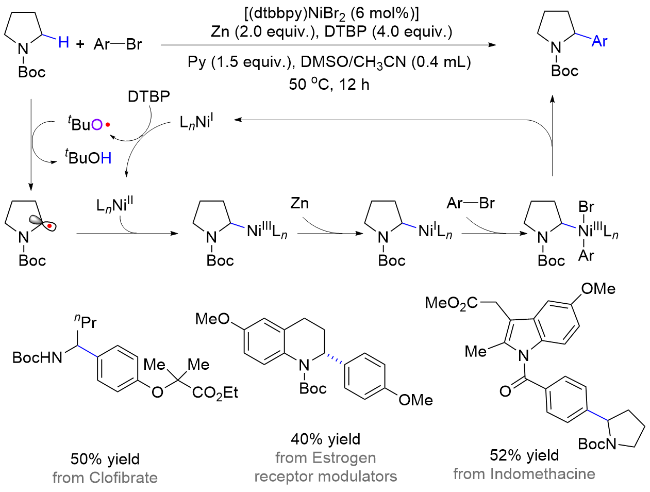
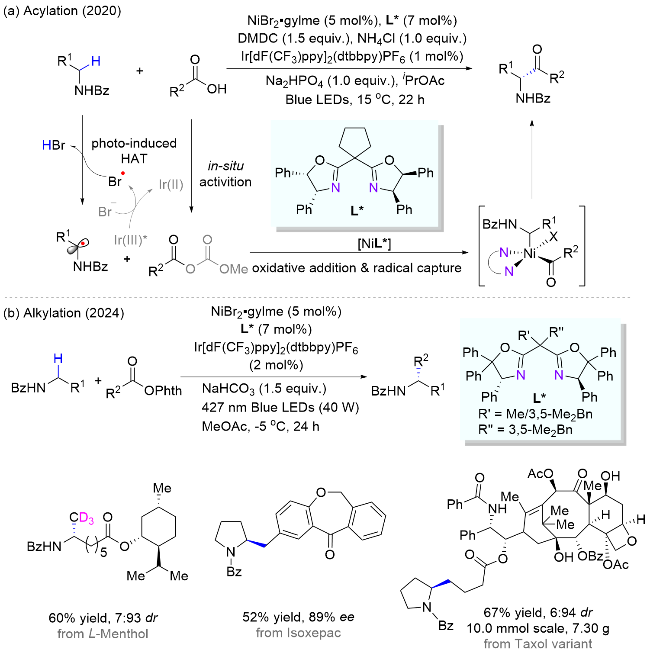
霍浩华课题组[36]发展了卤素自由基介导的不对称HAT催化策略. 2020年, 该课题组[37]采用简单的溴离子为HAT试剂, 完成了金属/光氧化还原催化的二级胺α-C(sp3)—H对映选择性酰化反应(Scheme 20, a). 首先, N-烷基苯并酰胺被光催化激发生成的溴自由基选择性攫氢形成一个前手性的氮α-碳自由基, 同时, 羧酸原位形成酰基亲电试剂; 其次, 手性镍催化剂与两个活性物种依次发生氧化加成和自由基捕获两个基元步骤, 得到的有机金属中间体经过还原消除产生高对映选择性的α-氨基酮. 该方法具有底物适用范围广、官能团兼容性好和对映选择性高等优点, 并且, 此方法适用于药物的后期修饰, 已经被成功应用于青蒿琥酯(治疗疟疾药物, 60%收率, 95:5 dr)、苯扎贝特(降血脂药, 70%收率, 85% ee)等药物分子的后期酰基化反应, 高效地引入了手性氨基酮药效官能团. 2024年, 该课题组[38]又采用邻苯二甲酰基作为活化羧酸的试剂, 通过分离或原位生成氧化还原活性酯的方式, 羧酸化合物可以在类似光催化的条件下脱羧生成甲基(三氘代)或烷基自由基, 继而在镍作用下与氮α-碳自由基进行不对称偶联反应(Scheme 20, b). 该方法在药物化学和氘代化学中存在广泛的应用前景, 能够高效合成具有生物活性的手性胺类化合物, 特别是那些含有能显著提高药物的生物活性和代谢稳定性的“魔法甲基”分子[30].
1.2.5 十钨酸盐介导
一般而言, 过渡金属催化自由基介导的C(sp3)—C(sp3)偶联反应需要形成高价的双烷基金属中间体, 然后经过选择性还原消除得到目标化合物. 然而, 该类金属活性中间体易于发生β-H消除、碳金属键断裂或者经历其它途径的还原消除过程, 同时自由基也易于发生自偶联副反应, 导致反应效率降低. 另外, 由于位阻影响叔碳中心, 与过渡金属之间往往难以形成相应的碳-金属键, 使得直接构建空间拥挤的季碳中心相对困难[41]. 基于上述问题, MacMillan课题组[41b,42]借鉴生物体内的酶促双分子均裂取代(SH2)反应机制, 结合光氧化还原催化, 实现了自由基介导、SH2途径的C(sp3)—C(sp3)交叉偶联反应. 2023年, MacMillan课题组[43]把该策略应用于含氮药物分子α-C—H键的后期甲基化反应中(Scheme 22). 使用甲酸衍生的活性酯为甲基化试剂, 十钨酸盐为α位C—H攫氢试剂和光氧化还原催化剂, 借助镍催化剂对相对不稳定的甲基自由基选择性捕获, 降低了溶液中游离甲基自由基的浓度, 使之选择性地与氮α-碳自由基发生偶联反应(而不是发生自偶联或者攫氢过程生成甲烷), 从而在温和条件下构建了一系列重要药物分子的甲基化衍生物, 证明了反应的实用性. 更重要的是, 该体系基于HAT-SH2双重催化机制, 能顺利构建复杂分子的sp3季碳中心.
2 氮β位C—H键官能团化
β位取代的胺类化合物是一类重要的有机骨架结构, 然而, 由于氮原子特殊的性质使得α位C—H键较为活泼, 不管是pKa值还是BDE值都要更低, 导致相邻的β位C—H键受到干扰, 其直接转化相对困难[44]. 因此, 与α位或者其他远端位置(如γ, δ)相比, HAT介导的β位的官能团化研究较少, 成功的例子不多.
2.1 分子内攫氢
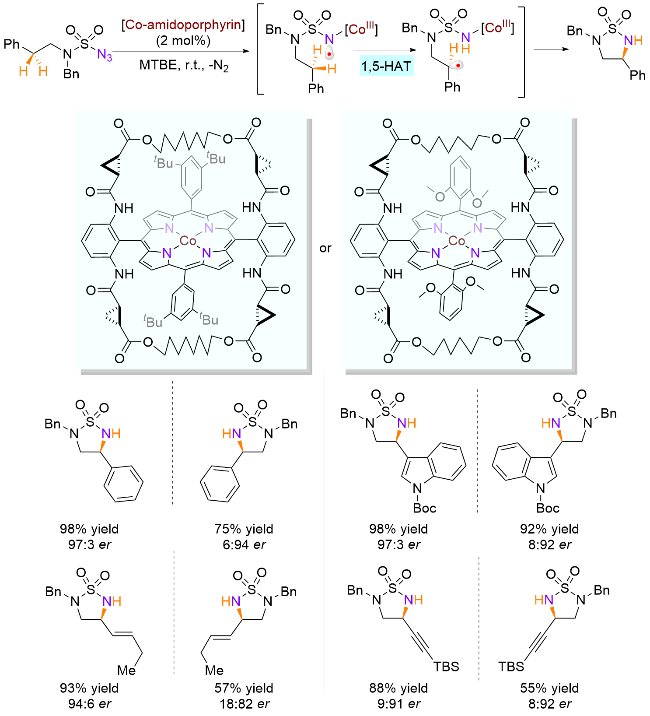
分子内氮原子辅助的自由基迁移受到迁移过渡态 C—H—N环张力以及迁移距离的影响, 一般优先发生1,5-HAT过程, 所以利用分子内氮自由基的攫氢过程一般得到的是δ (C-5)位官能团化产物. 不过, 通过引入额外含氮基团延长迁移路线, 利用分子内优势的1,5-HAT策略也可以实现氮β位C—H键的官能团化. 2019年, Zhang课题组[45]成功开发了新的Co(II)催化体系, 基于磺酰叠氮化物的叠氮基团脱去一分子氮气, 与手性卟啉钴催化剂生成N-Co(III)自由基中间体, 然后钴氮上的自由基经过1,5-HAT产生氮β-碳自由基, 最后胺化得到高对映选择性的环磺胺化合物(Scheme 23). 值得一提的是, 与之前发现的过渡金属催化自由基不对称反应不同, 该体系的对映选择性决定步骤为1,5-HAT过程, 即手性N-Co(III)中间体因为立体环境的限制, 会选择性地攫取亚甲基某一个C—H键, 生成瞬态构象约束的亚甲基自由基. 由于该自由基胺化速率快于消旋化, 所以得到的是手性保持的环胺化合物. 基于该过程, 作者通过调整手性卟啉钴的手性结构, 可以分别实现两种构型的构建.
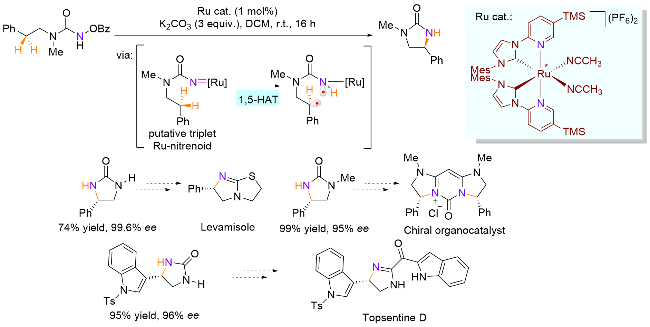
金属氮宾中间体是一类重要的活性中间体, 广泛应用于C—H键的插入反应中. 其中, 三线态金属氮宾一般表现为双自由基[46], 也同样具有发生分子内1,5-HAT的可能性 在这一方面, Meggers课题组[47]做了很多开创性的工作, 尤其是在手性过渡金属催化的不对称构建手性含氮分子领域. 2020年, 该课题组[48]合成了钌金属手性八面体配合物, 并成功将其应用于C(sp3)—H键的不对称环胺化反应(Scheme 24). 作者提出了相应的反应机理, 即钌和底物接触生成三线态的金属氮宾, 接着一个自由基经过1,5-HAT攫取β位的C—H键, 生成亚甲基碳自由基, 最后碳和氮自由基发生C—N键偶联, 得到目标化合物. 该反应具有优异的对映体过量值(最高99.6% ee), 这可能是由于配合物的手性中心在金属上, 更加接近底物的前手性中心, 因此有利于产物的对映选择性控制. 另外, 使用廉价金属铁催化, 消旋版本的环胺化也可以顺利进行[49].
2.2 分子间攫氢
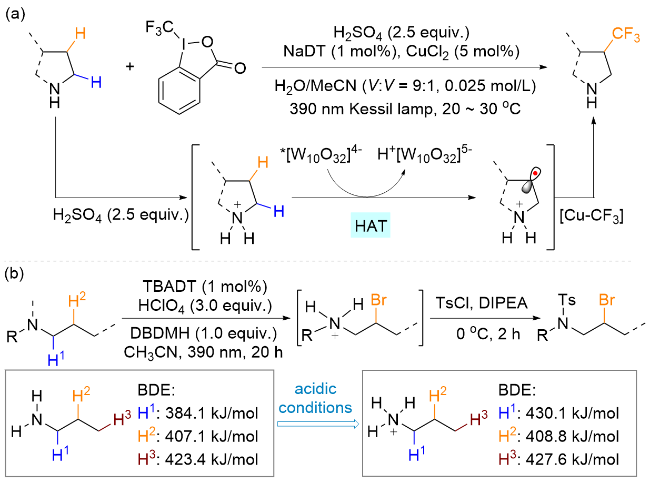
虽然由于氮原子的影响, α位的C—H键比β位活性更高, 但是利用分子内的1,5-HAT策略仍然可以克服该限制, 提高β位的选择性. 然而, 因为解离能的影响, 分子间的攫氢过程依然优先发生在α位, 使得基于自由基途径的β位C—H键选择性官能团化十分具有挑战性. 2020年, MacMillan 课题组[50]研究发现, 酸化后的铵盐氮原子会增加邻近α-C上氢原子的键能和降低其富电性, 从而不易被HAT试剂攫取, 此时电中性的铵盐β位C(sp3)—H键与激发态的[W10O32]4-试剂极性匹配, 因此优先被活化. 基于此发现, 研究人员实现了游离胺基β位的选择性三氟甲基化反应(Scheme 25, a). 更进一步, Martin课题组[51]在2024年基于DFT计算, 证明了胺基质子化后α-C—H键的解离能, 从之前的384.1 kJ/mol上升到430.1 kJ/mol, 而β位基本保持不变(408.8 kJ/ mol), 为酸性条件下HAT试剂选择性攫取氮β-C—H键提供了理论支持, 并以此实现了高β位选择性的溴化反应(Scheme 25, b). 不过, 上述策略的选择性受底物C—H键的性质影响较大, 优先反应在具有更低BDE值的 C—H键位置, 如叔碳原子上.
3 氮γ位C—H键官能团化
虽然分子内的自由基迁移倾向于以1,5-HAT的形式生成δ-碳自由基, 其区域选择性也并不是固定不变的, 影响分子内攫氢速率的因素有很多, 包括C—H键的性质(如3°、2°和1°碳或者苄位、烯丙基位等)以及辅助基团的类型. 研究显示, 虽然相同苄基底物的1,5-HAT (k1,5=1.3×105 s-1)比1,6-HAT (k1,6=1.5×106 s-1)大约快一个数量级[52], 但是通过引入一些含杂原子的辅助基团, 如磺酰基以及硅基可以使HAT优先发生1,6和1,7 位[53]. 基于此特点, 自由基形式的氮γ位C—H键官能团化一般是借助磺酰辅助基团实现的, 目前也仅局限于分子内攫氢过程.
3.1 氮氯/碘键引发
2020年, Roizen课题组[54]报道了自由基介导的磺酰胺类化合物γ位氯化反应(Scheme 26, a). 作者在外加磺酰胺基团氮原子上引入了氯原子形成N—Cl键作为氮自由基的前体, 通过光照引发和优势的1,6-HAT生成γ-碳自由基, 随后被氯原子捕捉得到目标产物. 同年, Muñiz课题组[55]开创性地报道了一种碘催化的选择性C(sp3)—H氟化反应(Scheme 26, b). 在该反应中, 磺酰胺的胺基可与单质碘反应原位形成N—I键, 接着在光照作用下产生氮自由基, 发生1,6-HAT后产生的γ-碳自由基再被碘自由基捕获生成碘代物. 与上述氯化过程不同的是, 碘代物继续被二芳基甲酸碘苯氧化, 从而生成三价烷基碘活性物种. 最后, 该物种与三乙胺氟化氢盐发生亲核氟化反应, 构建含氟化合物. 该方法无需金属参与, 仅需催化量的碘单质作为催化剂, 同时以廉价易得的三乙胺氟化氢盐为亲核氟化试剂, 在温和条件下成功合成了一系列氟取代的胺类化合物, 具有较高的应用价值.
3.2 氮钴键引发
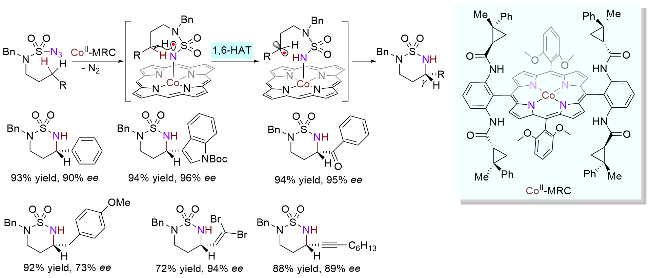
2018年, Zhang课题组[56]报道了基于手性卟啉Co(II)的金属自由基催化磺酰胺叠氮化物分子内1,6-HAT, 实现了不对称C(sp3)—H环胺化(Scheme 27). 作者曾报道了类似转化, 然而由于底物缺少γ位C—H键的竞争, 所以反应以1,5-HAT的形式进行, 实现了β位高选择性环胺化(Scheme 23). 与之相反, 在本研究中, 当底物同时存在β位和γ位C—H键时, 基于磺酰基的作用, 1,6-HAT优于1,5-HAT发生, 因此反应具有优异的γ位选择性. 该体系底物适用范围非常广泛, 像苄位、烯丙位、炔丙位和酰基α位, 都能顺利地以高对映选择性得到目标产物, 甚至与底物同时存在β、γ和δ位C—H键互相竞争时, 也不改变其选择性. 2020年, 基于碳自由基易于发生消旋化的机制, 该课题组[57]采用类似策略完成了氮γ位消旋的叔碳原子上C—H键立体汇聚式环胺化反应.
3.3 碳钴键引发
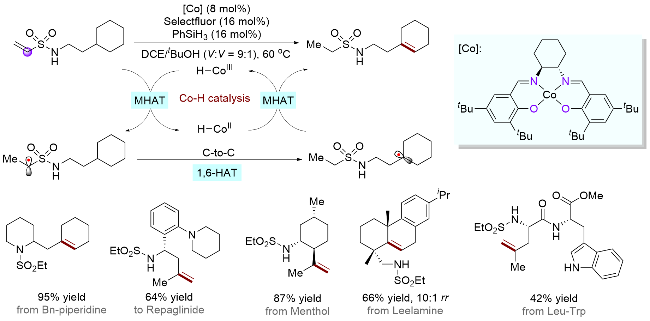
金属氢化物的氢原子转移(MHAT)策略通常应用于烯烃的氢官能团化和异构化反应中, 且伴随有碳金属键的均裂和碳中心自由基物种的生成. 2022年, Nagib课题组[58]结合MHAT和分子内HAT机制, 通过三重氢转移级联策略, 实现了胺类化合物远端及位置选择性的脱氢去饱和与中断MHAT的γ-胺碳自由基官能团化反应(Scheme 28). 作者首先在胺基上引入乙烯磺酰基辅助基团, 然后原位生成的钴氢物种与乙烯基发生第一重MHAT生成亚甲基自由基, 接着经历分子内碳原子到碳原子的第二重1,6-HAT, 生成γ-胺碳自由基, 最后发生第三重MHAT产生三价钴氢物种和对应的γ-δ去饱和高烯丙基胺产物, 显示出了强大的区域选择性. 最后, 辅助基团乙基磺酰基也可以通过简单的还原操作除去. 为了探究分子内HAT的选择性, 作者也进行了DFT计算, 结果显示, 基于辅助基团磺酰基的存在, 分子内同类型碳原子的1,6-HAT过渡态能量最低, 优先于1,5-和1,7-HAT过程, 这与过往研究结果一致[53].
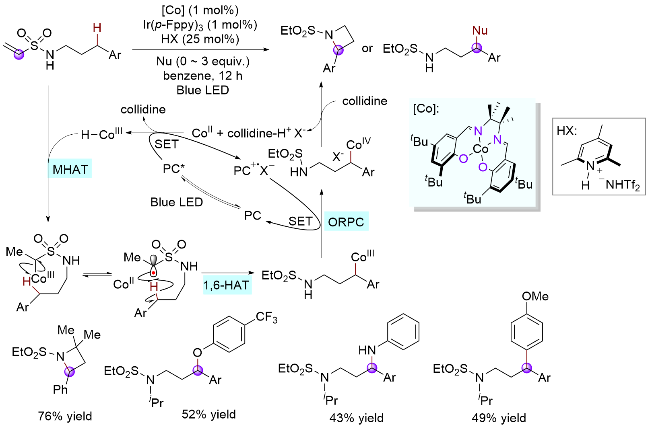
3.4 碳碘键引发
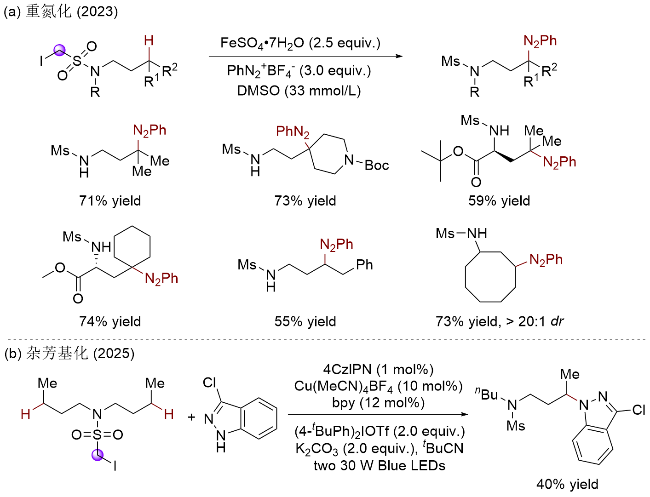
上述研究表明, 砜基α-碳自由基是一类高效的分子内HAT试剂, 可高选择性地实现磺酰胺氮γ-C(sp3)—H键的攫取, 辅助生成氮γ-碳自由基. 基于此, 吴新鑫课题组[60]探索了砜基α-碳自由基新的生成方式(Scheme 30, a). 其利用苯基自由基易于发生卤原子转移(XAT)过程均裂C—I键的特点, 实现了硫α-碳自由基的高效生成和分子内1,6-HAT. 其中, 苯基自由基来源于铁盐对苯基重氮盐的单电子还原过程, 同时该重氮盐也作为偶联试剂参与反应. 该体系可快速构建重氮化合物, 条件温和, 收率中等至优异(55%~90%). 最近该课题组[61]借助光/铜协同催化体系实现了区域选择性的C(sp3)—H键杂芳基化反应, 避免了使用等物质的量的过渡金属试剂, 使整个反应体系更加绿色高效(Scheme 30, b).
4 氮δ位C—H键官能团化
自由基介导的胺类化合物δ位官能团化最早于经典的Hofmann-Löffler-Freytag (HLF)反应中被发现, 具体为N—Cl化合物在强酸介质中紫外光解(或加热)产生氮自由基, 接着经历1,5-HAT攫取δ位C—H键生成碳自由基, 最后被氯原子捕获获得氯化产物. 该氯化物也可以在碱的作用下进一步与分子内胺基反应, 得到四氢吡咯类化合物. 然而, 由于容易生成氯化物, 此类反应的发展受到了限制, 大部分研究也只是在形成N—Cl键的方法上和均裂成氮自由基的方式上进行了探索.
4.1 氮氯键引发
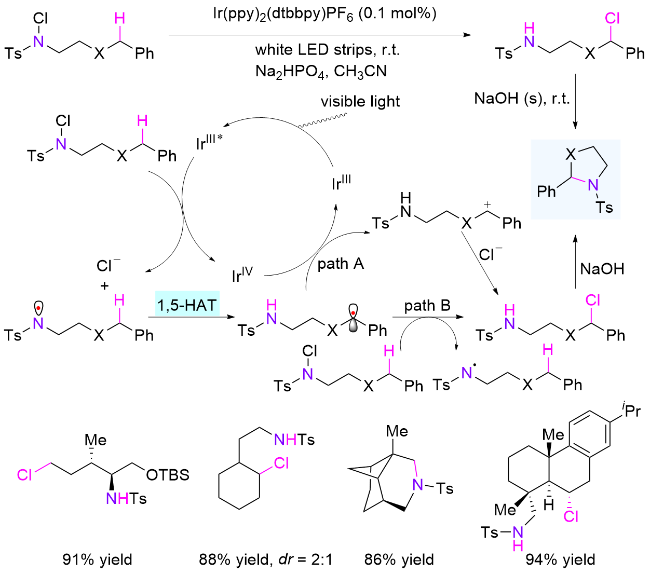
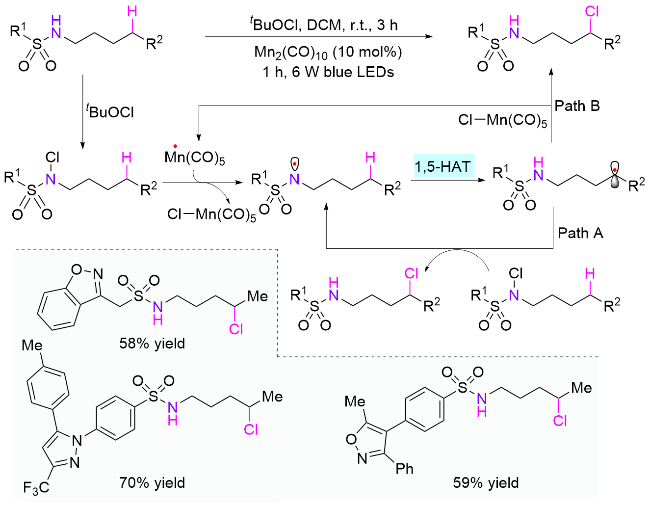
最近十年来, 通过改变反应条件和实验设备, HLF反应实现了更多类型的转化. 2015年, 俞寿云课题组[62]报道了使用Ir(ppy)2(dtbbpy)PF6作为光催化剂, 在弱碱性条件下实现磺酰胺的δ位C—H键氯化和分子内胺化反应(Scheme 31). 作者推测反应的氯化过程可能存在两种机制, 一种是产生的苄基自由基继续被光催化剂氧化得到苄基正离子, 继而与氯离子反应; 另一种是苄基自由基直接被另一分子的氯代磺酰胺氯化, 生成目标产物和氮自由基, 继续下一个循环, 即自由基链增长过程. 同一年, Muñiz课题组[63]也以碘为催化剂、二芳基碘苯为氧化剂, 原位生成碘代磺酰胺代替氯代磺酰胺, 在光照下完成了四氢吡咯结构的快速构建.
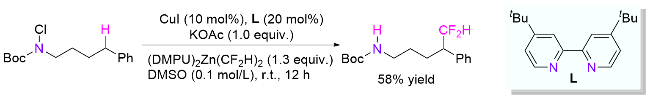
4.2 氮氟键引发
另一类突破HLF氯化反应限制的方法是使用氟代酰胺代替氯代酰胺. 由于溶剂效应减弱了氟原子的亲核性, 导致C—F键构建困难, 因此一般不会发生氟化过程. 借助该特性, 结合过渡金属催化, 胺类化合物δ- C—H键可以实现多种多样的转化.
2018年, Zhu课题组[67]报道了铜催化N-氟羧酰胺的远端δ位C(sp3)—H芳基化(Scheme 34, a). 与传统的HLF反应不同的是, 该研究使用一价铜还原氟代酰胺引发氮自由基, 然后经1,5-HAT得到碳自由基. 同时, 生成的二价铜物种F-CuIIX与芳基硼酸发生配体交换产生Ar-CuIIX. 最终, 碳自由基被Ar-CuIIX捕获生成三价铜物种, 经还原消除得到目标产物和一价铜, 一价铜继续参与催化循环. 类似地, Nagib课题组[68]也报道了铜催化氟代磺酰胺的δ位芳基化反应, 其中, 使用手性噁唑啉配体时最高可获得59%收率和65% ee值.
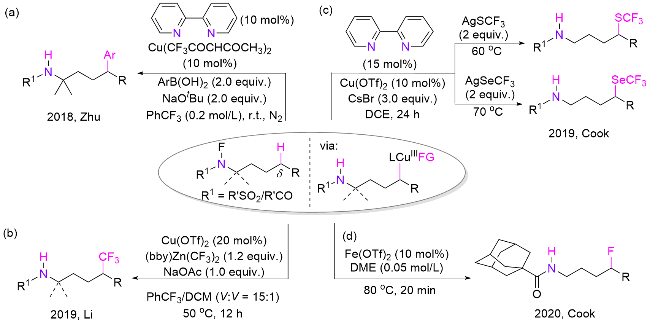
由于含氟基团在药物结构改造优化中的显著作用, 开发温和、高效的含氟化合物的合成策略十分重要. 借助过渡金属催化和1,5-HAT机制, 一系列含氟官能团可以顺利地被引入目标分子. 2019年, 李超忠课题组[69]报道了脂肪族C(sp3)—H键的直接三氟甲基化的方法(Scheme 34, b). N-氟磺酰胺和酰胺与Zn(CF3)2配合物在温和条件下反应得到相应的δ-三氟甲基化磺酰胺和酰胺类化合物. 同年, Cook课题组[70]发表了铜催化磺酰胺和酰胺衍生物的三氟甲硫基化(SCF3)和三氟甲硒基化(SeCF3), 分别使用相应的含氟银盐作为亲核试剂(Scheme 34, c). 虽然 C—F键构建困难, Cook课题组[71]借助特殊的金刚烷甲酰基作为辅助基团来提高迁移选择性和活性, 成功在氮δ位引入了氟原子(Scheme 34, d). DFT计算显示, 与传统HLF反应涉及的自由基链增长机理不同, 氟化反应很可能发生在三价氟化铁物种([F-FeIII])和碳自由基之间.
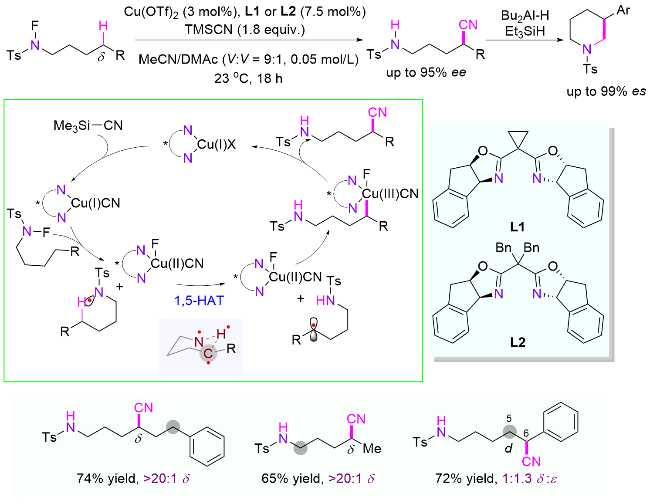
氟代酰胺δ位亚甲基C(sp3)—H键的直接官能团化会构建一个手性中心, 而配体辅助的铜催化体系为对映选择性控制提供了有效的途径. 2019年, Nagib课题组[72]使用手性双噁唑啉配体, 以氟代磺酰胺为底物, 在过渡金属铜催化下, 以56%~95%收率和86%~95% ee值得到高价值的手性δ-氰基胺化合物(Scheme 35). 同时, 该产物也可经一步转化为重要的手性β-芳基哌啶衍生物. 另外, 作者也进行了区域选择性研究, 通过不同位置 C—H键的氰基化产物比例分析发现, 选择性一般由优势的1,5-HAT决定. 不过, 控制实验表明底物结构也有着举足轻重的影响, 例如苄位相对高活性的C—H键相对提高了1,6-HAT的速率, 使得δ和ε位的产物比例为1:1.3. 同一时间, 王细胜课题组[73]也发表了类似的研究结果.
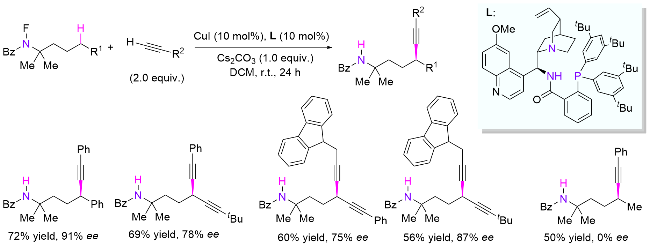
在氟代酰胺导向的远端不对称官能团化反应中, 原位生成的氮自由基可选择攫取羰基侧γ位或胺基侧δ位C(sp3)—H键. 因此, 通过底物设计, 同时借助过渡金属催化或酶催化完成同一体系不同位置手性中心的构 建[75]. 其中, 陈芬儿课题组[75a]在2024年报道了使用铜催化体系, 以氟代酰胺和易得的甘氨酸酯为底物, 分别实现了酰胺γ位和氮δ位不对称C(sp3)—H键烷基化, 构建了一系列对映体纯的α-氨基酸和多肽衍生物(Scheme 37). 该体系条件温和, 底物普适性广, 同时兼容一级、二级、三级和α-氧的C(sp3)—H键, 且具有优异的区域、对映和非对映选择性. 作者推测, 喹啉胺基配位的三价铜物种经历了分子内氧化产生氮自由基阳离子中间体, 随后去质子化生成关键的α-碳甘氨酸自由基物种, 最后与CuII发生分子内重组和不对称还原消除生成目标产物. 作者通过自由基捕捉实验检测到了甘氨酸同源偶联产物, 表明了甘氨酸自由基的存在, 从而证明了上述机理的合理性.
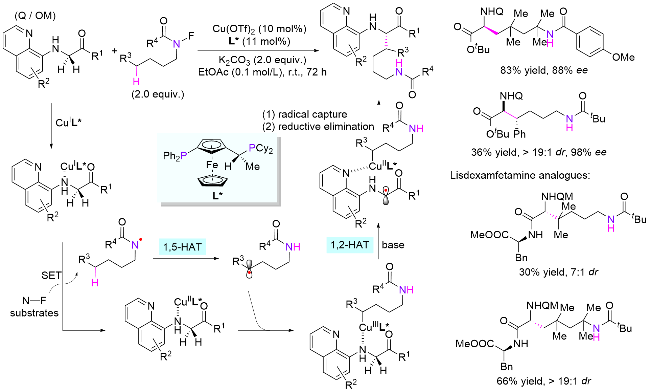
4.3 氮氧键引发
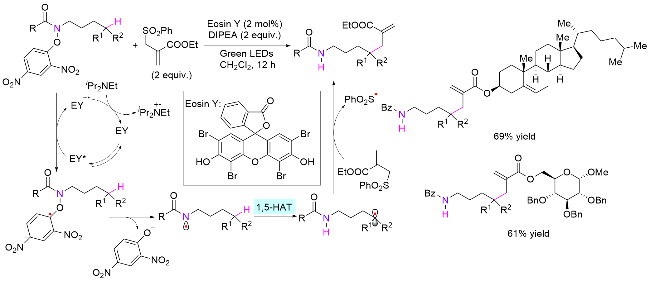
同年, 俞寿云课题组[78]也报道了光催化酰胺氮氧键导向的氮δ位C(sp3)—H键杂芳基化反应(Scheme 39, a). 反应体系具有条件温和及无金属参与等优势. 值得一提的是, 反应所使用的杂芳环偶联试剂无需预官能化, 同时拥有优异的区域选择性. 作者也进行了DFT计算, 证明通过Fukui函数模型可预测杂芳环反应的位点选择性和反应活性, 并且理论计算得到的预测结果和实验一致. 2020年, 该课题组[79]使用光/铜协同催化体系, 探索了氮氧键介导的酰胺δ位不对称氰基化反应(Scheme 39, b). 对于处于苄位的目标C—H键具有优异的收率和对映选择性(86%收率, 89% ee), 而针对一般亚甲基, 该噁唑啉配位的铜催化体系效果较差(80%收率, 12% ee).
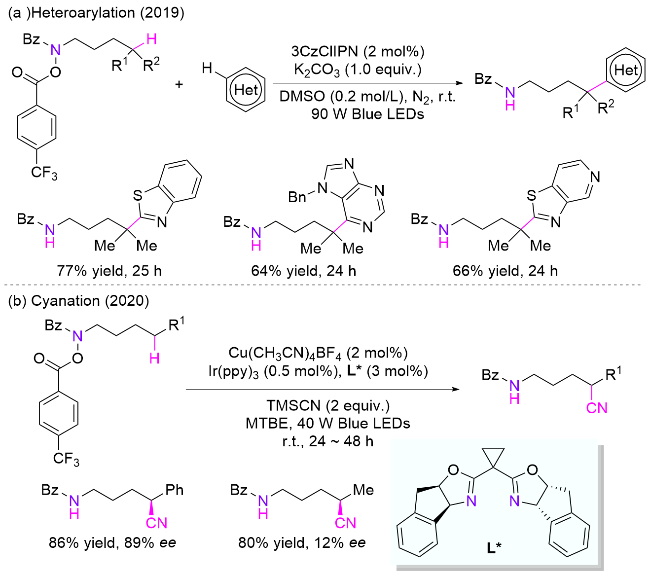
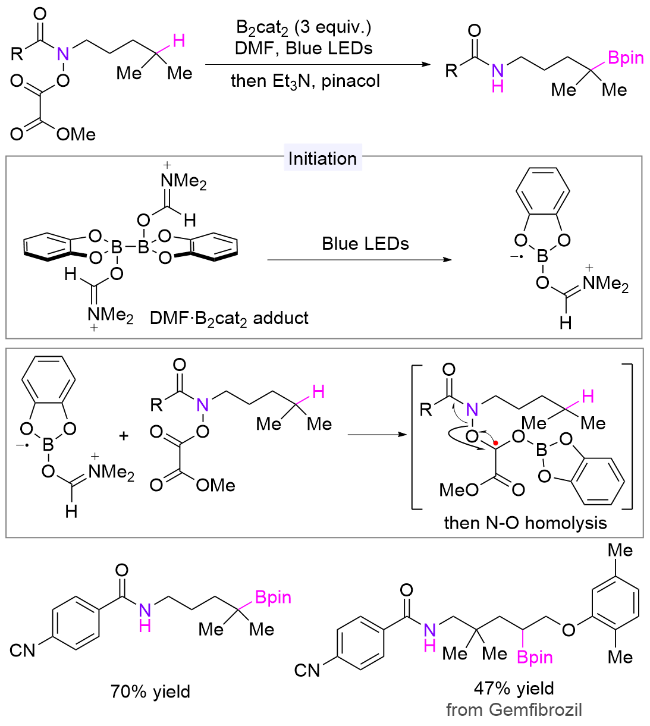
4.4 氮硫键引发
N-磺酰胺取代的酰胺结构是一类易于合成、稳定且可长期储存的化合物, 同时也是一类理想的氮自由基前体, 早前用于合成内酰胺[81]. 2018年, Studer课题组[82]在酰胺氮上引入N-烯丙基磺酰胺辅助基团, 并使用催化量的过氧化物引发自由基的产生和链传递, 通过简单改变砜试剂上取代基的种类, 分别实现了氮δ位C—N、C—Cl、C—Br、C—SCF3、C—SPh和C—C键的构建(Scheme 41). 该方法适用于未活化的三级和二级C—H键, 以及活化的一级C—H键的官能团化, 展示了广泛的底物适用性, 并可用于复杂生物活性分子的后期官能团化, 表明其在药物化学中也具有潜在应用价值. 以叠氮化反应为例子, 作者推测的机理为: 过氧化物在加热条件下分解为碳自由基, 该自由基与三氟甲磺酰基叠氮化物作用产生烷基叠氮化物、二氧化硫和三氟甲基自由基; 然后三氟甲基自由基加成到辅助基团烯丙基磺酰基上, 脱去一分子1,1,1-三氟丁烯后生成磺酰自由基; 接着该自由基脱除SO2并产生酰胺氮自由基, 后者经历分子内的1,5-HAT生成碳自由基; 最后, 该自由基被叠氮试剂捕获, 生成目标产物和三氟甲基自由基, 三氟甲基自由基参与下一个催化循环.
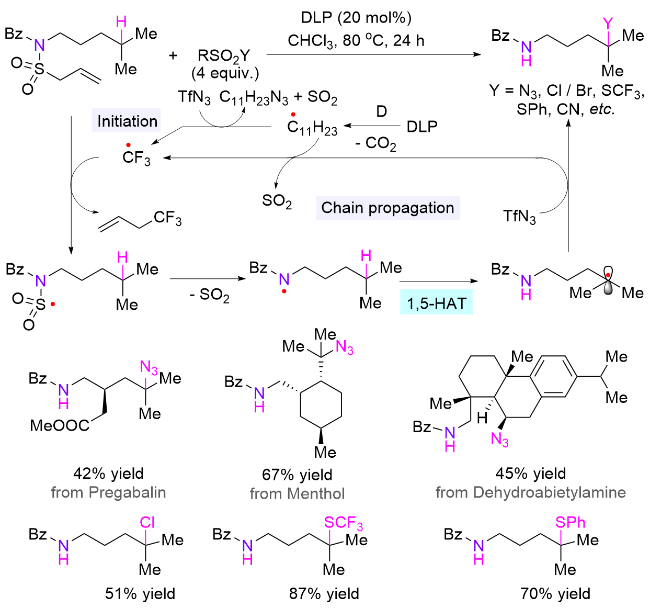
4.5 氮氢键引发
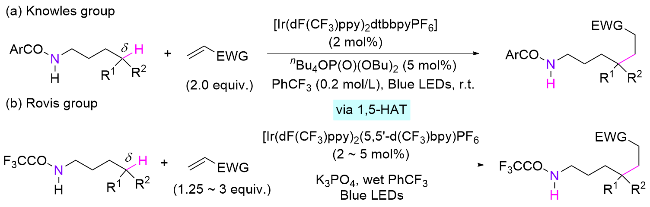
虽然通过在酰胺氮上引入基团或卤素的方式使氮自由基更容易生成, 其氧化型辅助基团的引入一般需要使用强氧化剂, 不仅增加了反应步骤和原料成本, 而且限制了底物适用范围. 因此, 如果能通过简单处理或直接激发N—H键生成氮自由基, 可以大大增加该类策略的应用前景. 然而, 一般酰胺N—H键的BDE值较高, 难以直接均裂生成氮自由基(≈418 kJ/mol)[83]. 2016年, Knowles课题组和Rovis课题组分别同时开发了类似的策略, 报道了光催化引发酰胺NH基团原位生成氮自由基的反应, 从而解决了均裂酰胺N—H键的难点, 实现了δ-C—H键与烯烃加成构建C(sp3)—C(sp3)键(Scheme 42). 两人都不约而同地使用碱活化胺基氢的方法, 其中, Knowles课题组[84]提出了协同“质子偶合电子转移(PCET)”的策略, 即使用光催化剂/Brønsted碱(四丁基磷酸铵, 可与NH形成氢键复合物)组合来调节N—H键的解离能, 过程为电子往光催化剂转移的同时质子也往Brønsted碱转移, 最终实现N—H键均裂活化(Scheme 42, a). 类似地, Rovis课题组[85]则使用吸电子的三氟甲基基团增加酰胺N—H键的酸性, 使之可以被磷酸钾脱质子, 降低其还原电势(Ep=+0.77 V<E1/2red[*Ir(III)/Ir(II)] = +1.21 V), 从而使Ir催化剂能够氧化酰胺基阴离子产生氮自由基(Scheme 42, b). 值得一提的是, 该远端δ位C(sp3)—H键的烷基化也可以通过在此类体系中加入镍催化剂和溴代烷实现[86].
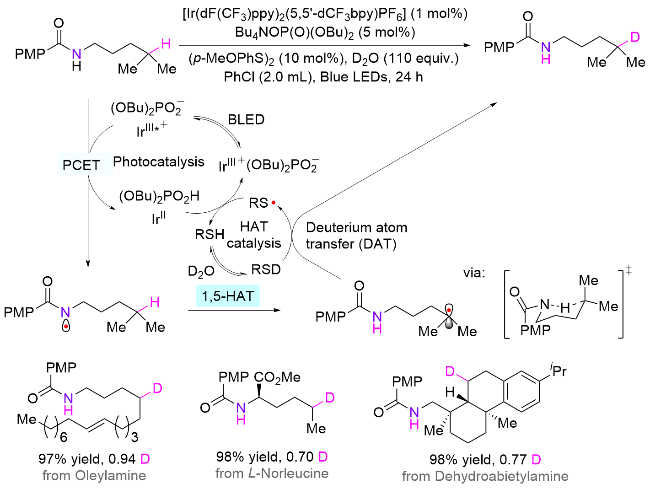
借鉴Knowles课题组发展的体系, 并结合硫醇参与的HAT催化体系, 谢劲课题组[87]于2022年报道了一例胺类化合物δ-C(sp3)—H键选择性氘化反应(Scheme 43). 在开始条件筛选之前, 作者首先对不同酰胺辅助基团下N—H键的解离能和氮自由基的半占轨道(SOMO)能量进行了计算. 结果显示, 对甲氧基苯甲酰胺底物拥有中等的BDE值和最高的SOMO能量(-7.58 eV). 依据极性匹配原则, 该辅助基可能会经历更快的1,5-HAT过程, 因此使用此基团辅助实现N—H键的均裂. 在对甲氧基苯甲酰基的辅助下, 大部分底物都能获得优异的收率和氘代率. 对于拥有多个δ位点的底物, 其多氘取代也能获得不错的结果. 由于强碱叔丁醇钾的加入对该反应无效, 因此排除了脱质子-电子转移机制, 并提出了如下机理: 首先IrIII和Bu4NOP(O)(OBu)2形成非共价的配合物, 接着该物种被光照激发至IrIII*, 此时底物的N—H键和(BuO)2PO2-形成氢键, 继而发生PCET过程产生氮自由基, 然后经1,5-HAT生成碳自由基. 由于大大过量的氘水存在, 硫醇上的氢被氘取代, 因此以RSD的形式存在. 亲核性的碳自由基与RSD发生氘原子转移(DAT), 从而生成目标产物和硫自由基, 后者可被二价铱还原, 同时从(OBu)2PO2H获得质子, 最终生成硫醇, 继续参与HAT催化循环.
2024年, 李坚军课题组[88]报道了光/钴协同催化的胺类化合物直接1,5-HAT, 实现δ位杂芳基化反应(Scheme 44). 与之前报道的氧化PCET机制不同的是, 作者发展了一种新的生成氮自由基的机制, 即光诱导分步的能量转移/质子转移(ET/PT)过程. 首先磺酰胺底物在光诱导下生成胺基自由基阳离子, 紧接着在Brønsted碱的辅助下脱去一分子质子(PT), 生成氮自由基; 其中, 六氟异丙醇与磺酰基形成的氢键作用可以稳定氮自由基, 从而提高反应收率; 然后氮自由基经历分子内1,5-HAT, 得到远端δ-碳自由基; 最后, 碳自由基和质子化的杂芳环发生加成反应, 再由钴催化剂[CoII]夺取氢原子生成目标产物和[CoIII-H]物种, 而后者可与质子转移步骤产生的质子反应生成氢气. 为了证明反应经历了自由基阳离子历程, 研究人员合成了N-甲基N-烷基磺酰胺类底物. 在标准条件下, 检测到了α位杂芳基化的产物, 说明该反应经历了叔胺自由基阳离子脱除α-H生成α胺甲基自由基过程. 另外, 重排反应结果也表明了α胺甲基自由基的生成, 反过来证明了在标准条件下胺基自由基阳离子的存在. 该方法具有广泛的底物适用性、高选择性和高效性, 并且在天然产物和药物分子衍生物的后期修饰中展示了巨大的应用潜力. 实验验证和理论计算进一步支持了提出的反应机制, 为该领域的进一步发展提供了新的思路.
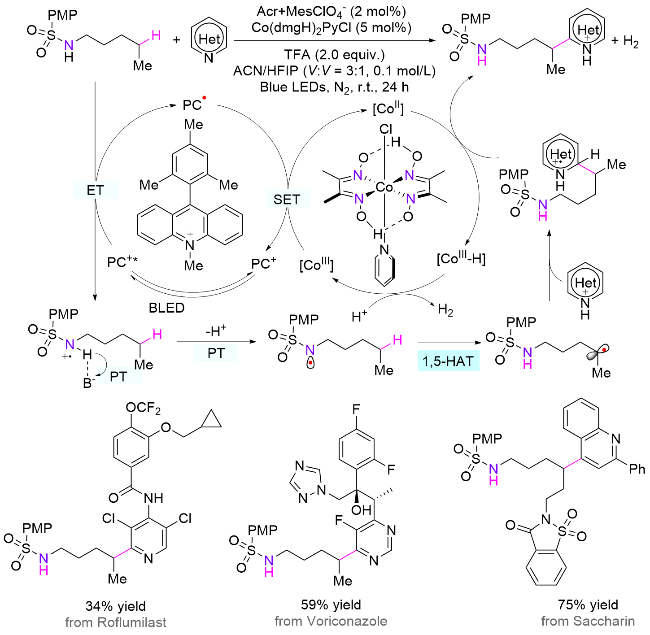
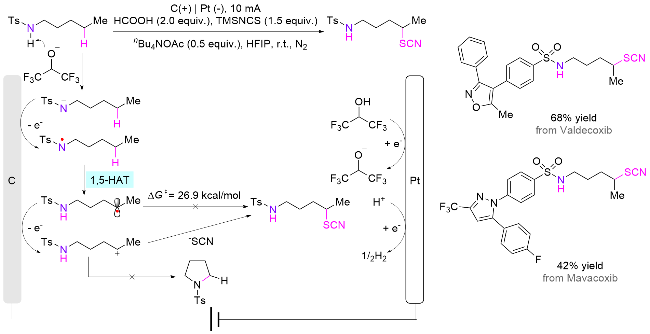
除了使用光催化体系, 电化学体系也可以直接激发生成氮自由基. 在该类体系中, 无需加入催化剂和氧化剂, 即可实现一般胺类化合物远端C—H键的官能团 化[89]. 2024年, 郭维斯课题组[90]报道了一例电化学促进磺酰胺基团辅助的胺类化合物远端δ位C(sp3)—H键硫氰化(Scheme 45). 在机理上, 与光催化体系类似, 首先需要借助六氟异丙醇阴离子脱除磺酰胺氮上的氢, 然后酰胺阴离子在阳极发生单电子氧化生成自由基, 接着经历1,5-HAT生成碳自由基. 该物种在阳极发生自由基极性交叉过程得到碳正离子, 最后与硫氰酸根阴离子反应生成目标产物. DFT计算表明, 碳正离子的硫氰化是热力学有利的产物.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 总结与展望
综上所述, 近十年来, HAT介导的胺类化合物C(sp3)—H键官能团化取得了巨大的进展. 该类策略通过自由基中间体的形成和到目标碳原子的转移, 可以实现特定C—H键的选择性活化. 同时, 反应一般在温和条件下进行, 可以有效避免传统方法中常见的副反应, 且具备较好的区域选择性. 经过多年发展, 多种高效催化剂(如光催化剂、金属配合物和HAT试剂)被开发出来,显著提升了HAT过程的效率和选择性. 特别是光催化HAT反应, 因其温和的反应条件和高效的能量利用而成为了研究热点. 在本文中, 我们按照胺基α、β、γ和δ位C—H键的选择性官能团化进行分类综述. 其中, α位由于氮原子的影响而具有较高的活性, 因此取得了广泛的发展, 尤其是分子间HAT催化体系进展迅速, 已成为高效合成复杂含氮分子的重要方法, 而其他β, γ和δ位的选择性官能团化可以通过设计引入合适的辅助基团, 借助优势的分子内1,5/1,6-HAT实现. 这些发展的策略和方法在药物合成、天然产物全合成和材料科学等领域展现出广阔的应用前景, 特别是为复杂分子的构建提供了一种高效、绿色的合成方法.
尽管基于HAT过程的胺类化合物选择性C—H键转化反应取得了显著进展, 但仍存在一些挑战和未来研究方向: (1)相比于其他位置, β位的转化在距离上处于不利位置, 成功的例子相对较少. 这是由于分子内的攫氢过程面临着α和γ位的竞争, 而分子间的自由基攫氢更是主要发生在α位, 虽然目前已有少量的例子通过酸化胺基的方式活化β位, 但其底物适用性不够理想, 因此需要开发更多普适性的策略. (2)目前胺类化合物选择性官能团化研究主要专注于胺基的同一碳链, 而仲胺和叔胺具有多个不同的烷基取代基, HAT试剂对不同侧链相似C—H键攫取的选择性控制模式研究不足, 还存在巨大的探索空间. (3)在分子内HAT反应中, 尽管通过辅助基团的设计可以实现胺基不同位置的转化, 而通过过渡金属和手性配体的结合可以实现立体选择性的控制, 但是配体对于区域选择性的控制作用却缺乏关注. 因此, 进一步研究基于配体控制的C—H键区域选择性官能团化新体系, 实现胺类化合物的高效转化, 是十分重要且具有挑战性的. (4)在分子内攫氢反应中, 一般需要使用氧化型辅助基团以便顺利发生单电子转移生成氮自由基, 而直接活化均裂胺基N—H键生成氮自由基来实现胺类化合物的选择性官能团化具有步骤和原子经济性等优势, 因此在近年来成为了研究重点. 然而, 现有的例子尚局限于δ位的转化. 如果该策略能成功应用到β和γ位C—H键的攫取, 不仅可以避免由于使用氧化型辅助基团导致的底物限制, 还可能大大拓展官能团化的种类和范围, 从而有助于解决远端β、γ和δ位相比于α位反应例子较少的问题, 促进胺类化合物选择性官能团化的进一步发展. (5) HAT策略可以在温和条件下活化惰性C(sp3)—H键, 并且优先攫取叔碳和仲碳上的氢原子, 继而形成一个手性中心, 因此在不对称催化反应中拥有巨大的应用潜力. 然而, 目前基于HAT过程的不对称反应大部分局限于形成苄位以及少量的烯丙位亚甲基手性中心, 而对两边不同烷基取代以及季碳手性中心的构建效果较差. 针对此类问题, 除了设计开发合适的手性配体, 使用手性HAT试剂实现对映选择性攫氢也是一种理想的解决策略[91].
总之, 基于HAT过程的胺类化合物选择性C—H键转化反应在有机合成领域具有重要的理论意义和应用价值. 通过不断优化反应条件、开发新型催化剂和深入的机理研究, 这一领域有望在未来取得更多突破性进展, 为有机合成化学和材料科学的发展做出重要贡献.
(Zhao, C.)