

甾体是一大类从植物、真菌、海洋生物和动物中分离得到结构多样的天然产物. 许多甾体具有广泛的药理和生理活性, 包括抗癌、抗炎和抗高血压等[1]. 依据其化学结构特征, 甾体可以分为经典甾体(classical steroid)和重排甾体(rearranged steroid)两大类. 经典甾体具有典型的[6-6-6-5]四环母核结构(图1A蓝色部分), 是甾体的主要组成部分. 重排甾体可以细分为三种类型[2]: (1)abeo-甾体(abeo-steroid), 通常是由经典甾体骨架发生最少一个C—C键迁移, 导致环系发生扩环或者缩环而形成的甾体(图1B红色部分), 但甾体的四环母核结构基本保持不变, 是重排甾体的主要组成部分; (2)开环甾体(seco-steroid), 通常是由经典甾体骨架发生最少一个环裂解而形成的甾体; (3)其它相关的三萜. 对经典甾体的持续研究最终促成了超过一百种获得美国食品药品监督管理局(FDA)批准上市的药物, 被广泛用于治疗癌症、炎症、疼痛和心力衰竭等疾病[3](图1A). 例如治疗过敏性鼻炎的糠酸氟替卡松(1)、治疗支气管哮喘的布地奈德(2)、治疗性腺机能减退的睾酮素(3)和治疗前列腺癌的醋酸阿比特龙(4). 经典甾体在化学药物体系中占有非常重要的地位, 仅次于抗生素的第二大类药物.
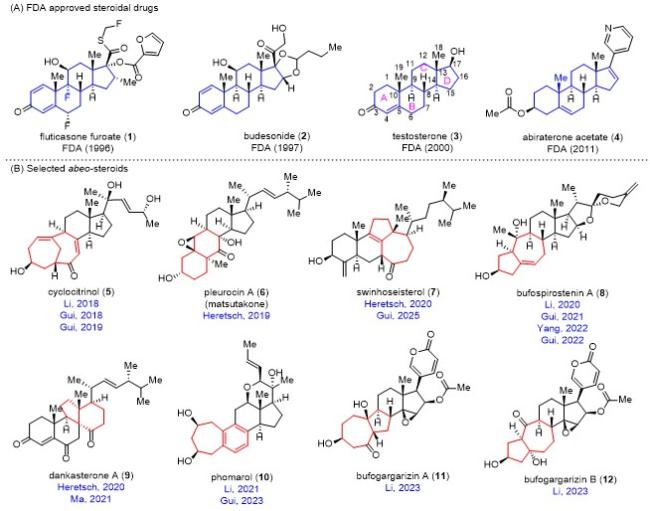
近年来, 越来越多的abeo-甾体被分离出来(图1B). abeo-甾体由于其新颖的化学结构和良好的生物活性, 吸引了全球合成化学家的关注[2]. 然而, 由于来源的稀缺性(植物提取产率低), 相比于被广泛和深入研究的经典甾体而言, 对abeo-甾体的研究仍然处于起步阶段. 从结构上看, abeo-甾体复杂且多变, 通常会含有一个中环体系(七元环)或者桥环体系, 且含有多个手性中心(图1B). 从合成上看, 由于结构的改变, 经典甾体的合成方法和策略(如多烯环化反应[4-6])很难直接用于abeo-甾体, 使得abeo-甾体的全合成具有很高的挑战性. 考虑到在经典甾体的成药性上取得的巨大成功, 对abeo-甾体的全合成研究, 一方面能够在构建新颖结构的有机合成方法上取得突破, 另一方面也能够从中获取稀缺的天然产物用于满足后续的创新药物研发, 为创新药的创制提供潜在的机遇, 具有非常重要的科学意义和应用前景.
目前, 文献中关于abeo-甾体的全合成领域的综述非常少, 只有Heretsch课题组[2]于2019年对重排甾体领域进行的首次综述, 以及李闯创课题组[7]和桂敬汉课题组[8]分别于2023年和2024年对自己课题组在abeo-甾体及相关领域的全合成进行的评述. 然而, 这三篇综述都没有对abeo-甾体的全合成研究进展进行全面和系统的综述. 需要指出的是, 自李闯创课题组[9]于2018年完成首个abeo-甾体cyclocitrinol的不对称全合成以来, 合成化学家对abeo-甾体的全合成研究取得了爆发式的重要进展. 因此, 对abeo-甾体的全合成研究进行一次系统的综述, 对该领域的重要进展进行一次梳理和归纳总结, 显得十分必要和及时. 本综述将根据时间顺序, 对近8年来(文献涵盖时间从2018年4月至2025年5月)报道的所有abeo-甾体全合成研究工作进行总结. 为方便读者对照, 将不同研究团队对同一个分子的全合成工作放在一起, 按照发表时间顺序逐一进行归纳和讨论, 重点阐述创新的合成策略和方法, 以期对相关领域的研究人员提供参考. 由于篇幅所限, 开环甾体[10-13]和一些兼具abeo-甾体和开环甾体结构特征的分子[14], 不在本综述的范围.
1 李闯创课题组不对称全合成cyclocitrinol
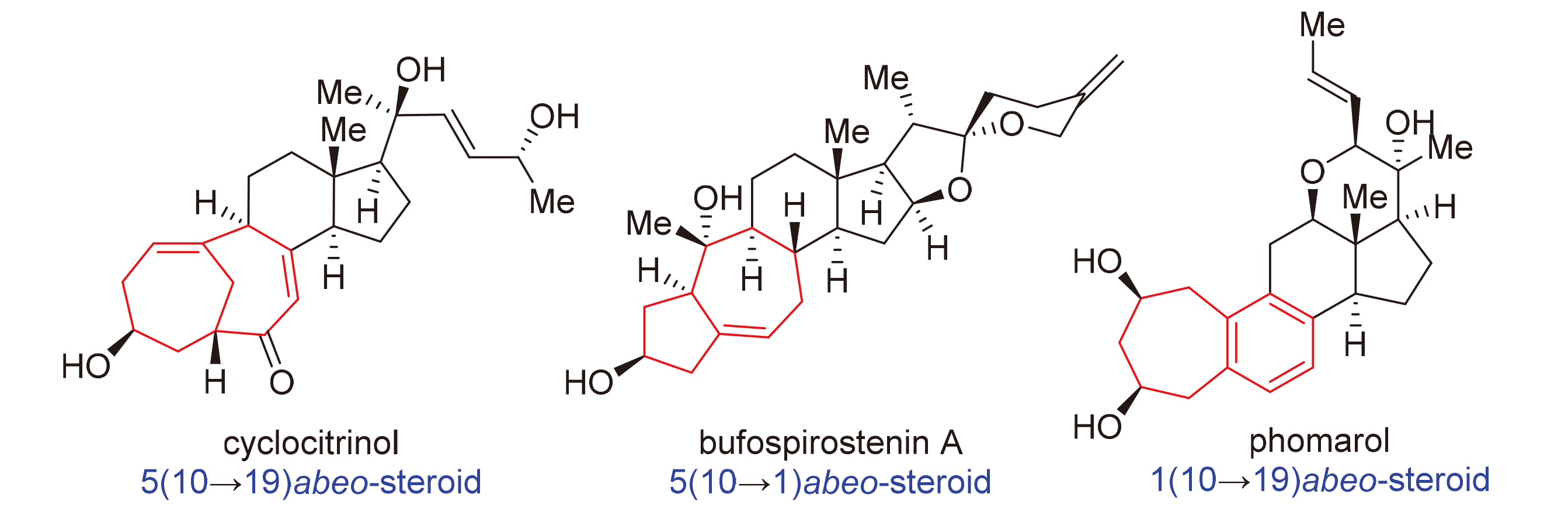
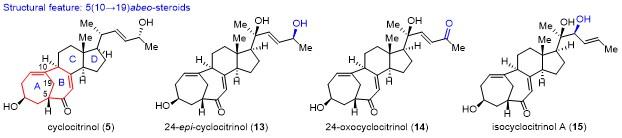
从结构上看, cyclocitrinol属于5(10→19)abeo-甾体天然产物. 该分子最早由Grafe等[15]从海绵青霉菌中分离得到, 后来, Crews等[16]对cyclocitrinol的结构进行了修订. 迄今为止, 已分离出超过25种不同的cyclo- citrinols家族天然产物(图2), 其中一些表现出广泛的生物活性[17]. 许多合成化学家先后投入到其全合成探索中, 如Schmalz课题组[18]以及Leighton课题组[19]等. 2018年, 李闯创课题组[9]以独创的type Ⅱ [5+2]环加成反应[20-21]为关键策略, 首次实现了cyclocitrinol的不对 称全合成.
cyclocitrinol的不对称全合成具体合成路线如Scheme 1所示. 以商业可得的化合物16为起始原料, 通过2步转化为烯酮, 接着发生碘代反应, 以3步56%的收率得到化合物17. 随后, 化合物17与18发生Stille偶联反应, 所得产物经1,4-还原, 以2步56%的收率得到化合物19. 化合物19与溴代物20衍生的锂试剂发生1,2-加成反应, 随后在四丁基氟化铵(TBAF)的作用下, 脱除两个叔丁基二甲基硅基(TBS)保护, 再在N-溴代琥珀酰亚胺(NBS)/水的作用下发生Achmatowicz氧化重排, 成功得到吡喃酮21. 化合物21经乙酰化, 随后在2,2,6,6-四甲基哌啶(TMP)作碱的条件下加热, 顺利发生type II [5+2]环加成反应, 成功构建了双环[4.4.1]桥环体系, 以4步56%的总产率得到22.
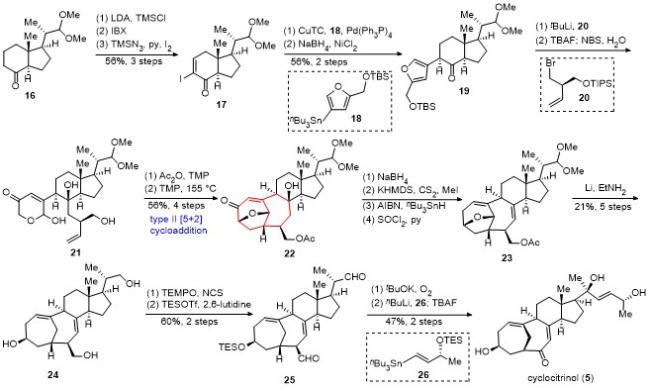
化合物22经硼氢化钠还原以及Barton-McCombie脱氧反应, 并在二氯亚砜和吡啶的存在下消除三级羟基, 得到化合物23. 化合物23在Li/EtNH2条件下选择性地还原切断氧桥, 与此同时缩醛被还原为醇, Ac保护基也被脱除, 以5步21%的总产率得到三醇化合物24. 利用2,2,6,6-四甲基哌啶氧化物(TEMPO)选择性地氧化化合物24的两个一级醇, 随后对二级醇进行三乙基硅基(TES)保护, 得到二醛化合物25. 化合物25在叔丁醇钾和氧气的作用下发生氧化去甲酰化反应, 得到相应的二酮化合物. 随后, 锡试剂26衍生的锂试剂对二酮化合物进行选择性的亲核加成, 引入侧链, 最后加入TBAF, “一锅法”脱除TES保护, 以2步47%的产率得到天然产物cyclocitrinol (5).
李闯创课题组以18步实现了cyclocitrinol的首次不对称全合成. 合成亮点主要包括: (1)基于课题组独创的type Ⅱ [5+2]环加成反应, 高效构建了双环[4.4.1]桥环体系, 这也是type Ⅱ [5+2]环加成反应在全合成中的首例应用; (2)采用温和的Li/EtNH2条件实现了七元氧桥的选择性还原切断反应.
2 桂敬汉课题组不对称合成cyclocitrinol及其它9个家族分子
2018年, 桂敬汉课题组[22]报道了第2例cyclo- citrinol的不对称合成方案, 具体的合成路线如Scheme 2所示. 以商业可得的孕烯醇酮(pregnenolone, 27)为原料, 通过2步简单反应得到环丙烷化合物28. 接着, 利用C(6)位羟基对C(19)位的甲基进行自由基介导的远程 C—H键官能团化, 并环化得到四氢呋喃化合物29, 其在酸性条件下发生环丙烷和四氢呋喃开环反应, 以2步49%的收率得到化合物30. 将化合物30的C(19)位羟基磷酸酯化, 并对C(7)位进行自由基溴代, 接着发生SN2取代反应, 以两步74%的收率得到硫醚化合物31. 化合物31经间氯过氧苯甲酸(m-CPBA)化学选择性的氧化得到亚砜32, 其在碱性条件下加热发生消除及仿生重排串联反应, 以2步54%的收率得到[4.4.1]桥环化合物33. 对化合物33的C(5)-C(6)位的双键进行选择性硼氢化氧化得到醇, 接着发生Jones氧化, 以74%的收率得到二酮化合物34. 最后, 通过烯基负离子对甲基酮立体选择性加成, 并脱除Ac保护, 以10步完成了cyclocitrinol (5)的合成.
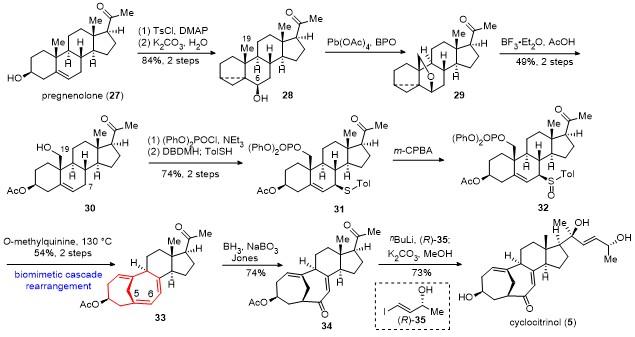
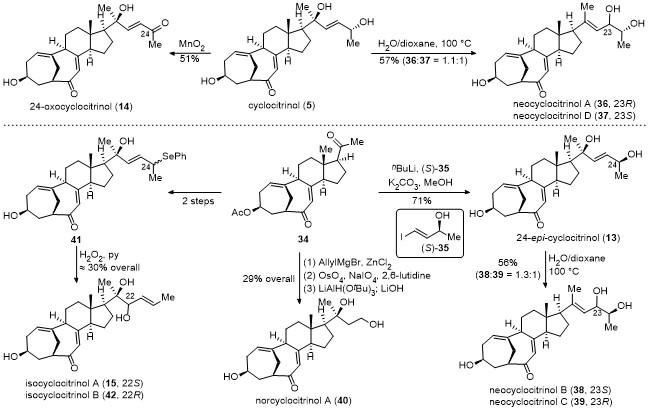
采用(S)-35与丁基锂形成的烯基负离子对化合物34的甲基酮进行立体选择性加成, 并脱除Ac保护, 以71%的收率得到24-epi-cyclocitrinol (13). 化合物13在以水和二氧六环作溶剂和加热的条件下, 同样发生1,3-烯丙醇重排反应, 以中等收率得到neocyclocitrinol B (38)和neocyclocitrinol C (39). 采用烯丙基格氏试剂对化合物34的甲基酮进行加成, 所得的端烯经氧化切断及还原, 最后脱除Ac保护, 以29%的总收率得到nor- cyclocitrinol A (40). 化合物34经过2步转化得到化合物41, 其在双氧水条件下发生seleno-Mislow-Evans重排反应, 得到isocyclocitrinol A (15)和isocyclocitrinol B (42).
桂敬汉课题组以10步反应完成了abeo-甾体cyclocitrinol的简洁高效仿生合成. 合成亮点主要包括: (1)基于仿生串联重排反应, 高效构建双环[4.4.1]桥环体系; (2)实现了10个cyclocitrinols的统一式高效合成, 并为该家族分子的生源合成假说提供了实验支持.
对于abeo-甾体cyclocitrinol分子, 李闯创课题组采用独创的type Ⅱ [5+2]环加成反应为关键策略, 首次实现了cyclocitrinol的不对称全合成; 而桂敬汉课题组通过巧妙的仿生串联重排反应策略, 完成了cyclocitrinol的简洁高效仿生合成.
3 Heretsch课题组不对称合成pleurocin A (matsutakone)和pleurocin B
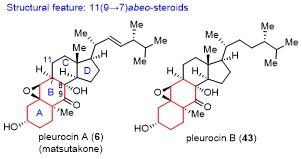
天然产物pleurocins A和B具体的合成路线如Scheme 4所示. 以已知的化合物44(由商业可得的麦角甾醇经3步58%的总收率转化而来)为原料, 在2-碘酰苯甲酸(IBX)和樟脑磺酸(CSA)条件下, 分别在C(9)-C(11)位和C(14)-C(15)位引入两个双键, 接着采用L-selec- tride, 选择性地进行共轭还原得到化合物45. 化合物45的双烯体首先与单线态氧(1O2)发生[4+2]环加成反应, 然后用LiAlH4还原切断过氧键, 并在酸性条件下发生消除反应, 以4步28%总收率得到二醇46. 二醇46经C(11)位羟基诱导的立体选择性的环氧化反应, 然后用Raney Ni进行插烯还原开环, 得到C(9)-C(11)位的顺式邻二醇, 后者用醋酸碘苯[PhI(OAc)2]处理, 氧化切断C环, 生成醛酮中间体, 并发生形式上的氧杂[4+2]环加成反应, 以2步56%的收率得到缩醛化合物47.
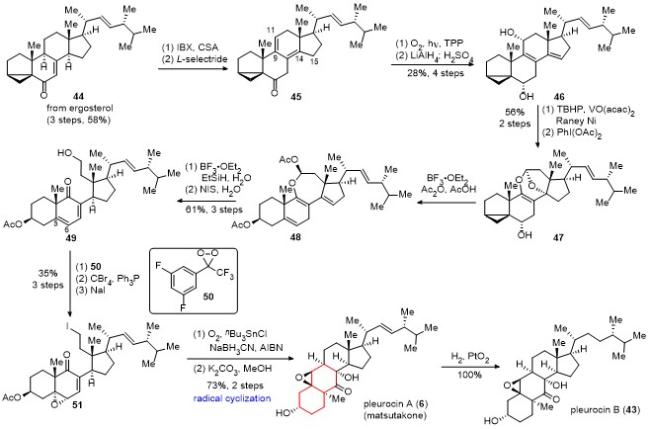
化合物47在酸性条件下选择性地打开四氢呋喃环, 得到Ac保护的缩醛化合物48, 后者在三氟化硼乙醚和三乙基硅烷条件下, 还原得到七元烯醚环, 并用N-碘代丁二酰亚胺(NIS)氧化, 以3步61%的收率得到开环化合物49. 采用过氧化合物50, 立体和区域选择性地对化合物49的C(5)-C(6)位双键进行环氧化, 接着对一级醇进行溴代, 最后将溴代物转化为碘代物51 (3步, 总收率35%). 化合物51在氧气氛围下, 顺利发生自由基环化反应, 经脱除Ac保护, 以2步73%的收率得到天然产物pleurocin A (matsutakone). 天然产物pleurocin A (6)经氢化反应, 可以定量地得到另一个天然产物pleurocin B (43).
Heretsch课题组完成了11(9→7) abeo甾体pleuro- cin A (matsutakone)和pleurocin B的高效合成. 合成亮点主要包括: (1)氧化切断C环, 并原位发生串联的形式上氧杂[4+2]环加成反应; (2)合成后期通过氧气淬灭的自由基环化反应重构C环.
4 Heretsch课题组不对称合成swinho- eisterol A, dankasterone A, dankasterone B和periconiastone A
具体的合成路线如Scheme 5所示. 以已知的化合物54为原料(由商业可得的麦角甾醇经4步42%的总收率转化而来), 在醋酸碘苯和碘单质条件下, 顺利发生自由基介导的骨架重排反应, 以76%的收率得到化合物55. 化合物55经还原脱碘、酸性条件下开环丙烷、碱性条件下脱除Ac保护和戴斯-马丁试剂(DMP)氧化, 最终以4步68%的收率得到dankasterone B (52). 52经Saegusa-Ito氧化, 以36%的收率得到天然产物danka- sterone A (9). 另外, 52在1,8-二氮杂双环[5.4.0]十一碳- 7-烯(DBU)条件下, 顺利发生分子内aldol反应, 以86%的收率得到periconiastone A (53).
化合物54在氧化汞和碘单质条件下, 顺利发生自由基介导的另一种路径的骨架重排反应, 以68%的收率得到化合物56. 化合物56经一锅法的1,6-还原和LiAlH4还原、臭氧选择性切断侧链的双键以及Julia- Kocienski烯基化反应, 以3步35%的收率得到含有顺式双键的化合物58. 化合物58在硼烷存在条件下, 对侧链的双键进行硼氢化氧化反应, 与此同时, 酮羰基也被还原为醇, 接着将1,3-二醇进行缩酮保护以及Barton- McCombie脱氧反应, 以4步54%的收率得到化合物59. 化合物59在酸性条件下脱除缩酮保护, 并打开环丙烷, 接着用二异丁基氢化铝(DIBAL)还原乙酰基, DMP氧化二醇为二酮, DBU介导双键移位, 最终以4步50%的收率得到烯酮60. 烯酮60经Luche还原所得的烯丙醇经过2步转化得到碘代物61. 化合物61经Stork自由基环化和Tamao氧化, 得到1,3-二醇, 其一级醇经三氟甲磺酸酯化和消除反应, 以2步44%的收率得到天然产物swinhoeisterol A (7).
Heretsch课题组通过条件可控的自由基骨架重排串联反应, 分别以9步、10步、10步、21步反应完成了四个abeo-甾体dankasterone B、dankasterone A、periconiastone A和swinhoeisterol A的简洁高效合成.
5 马志强课题组不对称全合成dankasterone A, dankasterone B和periconiastone A
2021年, 马志强课题组[32]通过金属催化的氢转移(MHAT)自由基环化策略, 完成了三个abeo-甾体dankasterone B、dankasterone A和periconiastone A不对称的全合成.
具体的合成路线如Scheme 6所示. 易得的手性化合物62经Wittig反应和溴代反应, 以2步66%的收率得到化合物63. 已知的手性二醇64经选择性地保护一级羟基, DMP氧化二级羟基, 所得的羰基先发生α-氢构型翻转得到顺式[5-6]并环, 接着发生三氟甲磺酸酯化和Stille偶联, 最后经DMP氧化, 以6步50%的总收率得到烯醛65. 化合物63与叔丁基锂进行锂卤交换, 所得锂试剂与烯醛65发生1,2-加成, 接着用DMP氧化, 以2步67%的收率得到烯酮化合物66. 经过条件优化, 关键反应化合物66的金属催化氢转移(MHAT)在67的催化下, 顺利发生环化, 以中等收率得到化合物68. 中间体68在Li/NH3条件下脱除Bn保护和还原烯酮, 再用LiAlH4还原得到二醇, 其经乙酰化和消除反应, 以4步32%的收率得到烯烃69. 化合物69经脱除缩酮保护、LiAlH4还原、Mitsunobu反应以及三氧化铬和3,5-DMP对烯丙基氧化, 最终以4步51%的收率得到烯酮70. 采 用Danieli等开发的条件, 经过自由基重排反应, 化合物70以2步44%的收率顺利转化为二酮71. 最后, 脱除化合物71的Ac保护, 氧化为醛酮化合物, 其经Julia- Kocienski烯基化反应, 以3步26%的收率得到天然产物dankasterone B (52). 52经脱氢反应, 以28%的收率得到天然产物dankasterone A (9), 并有部分原料回收. 另外, 52在DBU/三甲基硅基三氟甲磺酸酯(TMSOTf)条件下, 顺利发生分子内aldol反应, 以46%的收率得到perico- niastone A (53).
马志强课题组利用MHAT触发的自由基环化和仿生自由基重排以及分子内羟醛缩合为关键策略, 完成了dankasterone A, dankasterone B和periconiastone A的不对称全合成.
6 桂敬汉课题组不对称全合成swinhoei- sterols A~C
2025年, 桂敬汉课题组[33]以Negishi/Heck串联反应和Baran烯烃还原偶联反应为关键策略, 完成了abeo-甾体swinhoeisterols A~C不对称的全合成.
具体的合成路线如Scheme 7所示. 易得的手性化合物72经Li/NH3立体选择性地还原为酮、在酸性条件下脱除缩酮保护、Mukaiyama脱氢反应以及氯代反应, 以4步54%的收率得到烯酮74. 化合物75与正丁基锂制备为锂试剂, 然后对化合物74进行立体选择性的1,4-加成反应, 并原位转化为三氟甲磺酸酯76. 化合物76在Pd(OAc)2/PhCPhos条件下, 与锌试剂77顺利发生Negishi/Heck串联反应, 经脱除TBS保护和Ley氧化, 以3步37%的收率得到化合物78. 经过大量筛选发现, 将化合物78的醛基转化为氰基, 其在Fe(dpm)3条件下, 所期望的Baran烯烃还原偶联反应可以顺利发生, 所得产物经叔丁醇钾处理, 得到高非对映选择性[C(20) dr=7.7∶1]的环化产物. 该环化产物经DIBAL还原, 以4步32%的总产率得到化合物79. 需要指出的是, 化合物78在Fe(dibm)3条件下, Baran烯烃还原偶联反应也可以顺利发生, 以33%的收率得到79, 但非对映选择性较差[C(20) dr=1.3∶1], 且所得产物经DBU处理并不会提高非对映选择性, 反而会降低非对映选择性.
化合物79经DIBAL还原和引入侧链以及脱保护等4步简单转化以42%的总收率得到化合物81. 化合物81经过Grieco-Sharpless反应选择性对一级羟基进行消除, 以65%的收率得到天然产物swinhoeisterol A (7). 另外, 采用臭氧对化合物81的四取代双键进行环氧化, 再经过Grieco-Sharpless反应选择性对一级羟基进行消除, 以3步37%的收率得到天然产物swinhoeisterol C (83). 天然产物swinhoeisterol C (83) 经叔丁醇钾处理, 打开环氧, 以89%的收率得到天然产物swinhoeisterol B (84).
7 李闯创课题组不对称全合成Buforspiro- stenin A
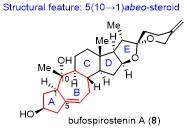
具体合成路线如Scheme 8所示. 该合成从Hajos- Parrish酮衍生的已知化合物85出发, 在氢化钠条件下选择性地和碘代物86在α位烷基化, 随后在硼氢化钠和NiCl2•6H2O体系中发生立体选择性1,4-还原, 采用CeCl3•7H2O脱除缩醛保护, 而后发生Seyferth-Gilbert增碳反应引入端炔, 以4步22%产率得到化合物88. 化合物88和联烯锂试剂发生1,2-加成反应, 顺利得到关键反应前体化合物89. 经过一系列的条件优化, 化合物89最终在[RhCl(CO)2]2为催化剂, 一氧化碳氛围和甲苯回流条件下, 发生分子内烷氧基联烯和炔的Pauson-Khand反应, 顺利得到[5-7-6-5]四环骨架化合物90. 接下来的任务主要集中在调整四环骨架的氧化态以及引入连续的多个手性中心.
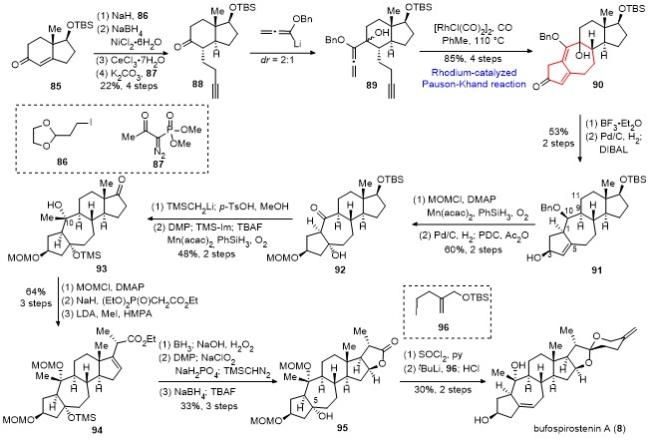
化合物90在三氟化硼乙醚的作用下选择性地消 除叔醇, 接着在Pd/C氢气条件下选择性地氢化C(9)-C(11)位和C(1)-C(10)位的两个双键, 在C(1)和C(10)位引入期望的手性中心, 最后经DIBAL还原, 以2步53%的收率得到化合物91. 化合物91首先经甲氧基甲基醚(MOM)保护羟基, 接着利用Mukaiyama水合反应引入C(5)的羟基, 再经脱除C(10)位的苄基以及重铬酸吡啶嗡盐(PDC)氧化, 得到化合物92. 随后利用Peterson烯化反应对化合物92引入环外双键, 脱除TBS保护并用DMP氧化为酮, 利用TMS保护C(5)的羟基和Mukaiyama水合反应引入C(10)位叔醇, 以2步48%的产率得到化合物93. 化合物93的C(10)位羟基先保护为MOM醚, 接着利用Horner-Wadsworth-Emmons反应引入酯基片段, 随后在二异丙基氨基锂(LDA)/六甲基磷酰三胺(HMPA)的条件下立体选择性地甲基化, 以3步64%的收率得到化合物94. 化合物94经过一系列的氧化还原操作, 包括硼氢化氧化、DMP氧化、Pinnick氧化、原位甲酯化、硼氢化钠还原、原位内酯化和“一锅法”加入TBAF脱除C(5)位的TMS保护基, 以3步33%产率得到化合物95. 化合物95在二氯亚砜和吡啶的条件下, 区域选择性地消除C(5)位叔醇, 随后和化合物96衍生的锂试剂发生1,2-加成反应, 最后在盐酸的条件下原位生成螺环, 同时脱除C(3)位和C(10)位的两个MOM保护基, 以2步30%的产率完成了bufospirostenin A的全合成.
8 桂敬汉课题组不对称合成buforspirostenin A和ophiopogonol A
2021年, 桂敬汉课题组[35]报道了2条bufospiro- stenin A简洁合成路线. 具体合成路线如Scheme 9所示. 该合成从tigogenin lactone的衍生物双烯酮97出发, 在醋酸和光照的条件下发生光促山道年重排反应, 将[6-6]环系重排为[5-7]环系, 得到化合物98, 但重排的产率仅为10%(克级别制备). 为了提高反应的产率, 通过碘代和插羰反应在C(2)位引入羧基, 该底物在醋酸和光照的条件下发生光促山道年重排反应, 其产率提高到58% (克级别制备), 最后经脱羧反应, 以4步28%的总产率得到化合物98. 化合物98经Ac保护和Luche还原, 随后与化合物99发生钴催化的氢硒化反应, 以3步61%的产率得到硒醚100. 化合物100经过苯硒溴引发的E2消除反应, 最后采用李闯创课题组螺环的构建策略, 以2步27%的产率得到天然产物bufospirostenin A.
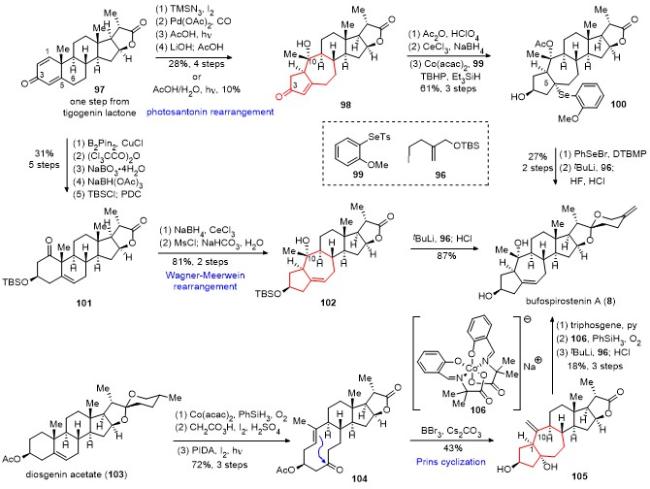
第二条合成路线同样采用化合物97为起始原料, 经1,4-加成反应在C(1)位引入硼酸频哪醇酯, 选择性地在C(5)-C(6)位生成双键得到共轭烯醇三氯乙酸酯, 采用过硼酸钠将C(1)位的硼酸频哪醇酯氧化为醇, 非对映选择性地还原C(3)位的酮羰基并用TBS保护, 最后用PDC氧化C(1)位的羟基, 以5步31%的总收率得到化合物101. 化合物101经Luche还原C(1)位酮羰基为羟基, 接着将羟基转换为甲磺酸酯, 得到Wagner-Meerwein重排前体, 其在碳酸氢钠作碱、四氢呋喃和水作溶剂的条件下, 原位发生Wagner-Meerwein重排, 以2步81%的收率得叔醇102. 最后采用相似的螺环构建方法, 以9步18%的总产率完成bufospirostenin A的高效合成.
2022年, 桂敬汉课题组[36]又报道了一条新的bufo- spirostenin A的简洁合成路线(Scheme 9). 该合成以diosgenin acetate(103)为原料, 采用Mukaiyama 水合反应引入C(5)位的羟基, 随后在过氧乙酸、碘和硫酸的条件下对螺环进行氧化降解, 得到对应的内酯. 该内酯在醋酸碘苯和碘的条件下, 经过光照, 发生C(5)位羟基介导的自由基β-碎裂化反应, 以3步72%的产率得到十元环化合物104, 其双键为单一反式构型. 经过大量的条件筛选, 发现在三溴化硼和碳酸铯的条件下, 跨环Prins环化反应顺利发生, 以43%的收率得到顺式[5-7]并环化合物105. 化合物105在三光气和吡啶的条件下消除叔醇, 随后利用Carreira钴催化剂(106)催化的Mukaiyama水合反应引入C(10)的叔醇, 最后采用相似的螺环构建方法, 以7步完成了bufospirostenin A的合成.
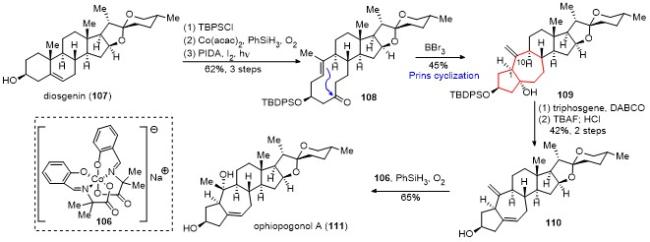
与此同时, 基于跨环Prins反应策略, 桂敬汉课题 组[36]也完成了ophiopogonol A[37](111)的合成(Scheme 10). 该合成以diosgenin(107)为原料, 先将羟基用TBDPS保护, 接着采用Mukaiyama水合反应引入C(5)位的羟基, 随后在醋酸碘苯和碘的条件下, 经过光照, 发生C(5)位羟基介导的自由基β-碎裂化反应, 以3步62%的产率得到十元环化合物108, 其双键为单一反式构型. 在三溴化硼的条件下, 跨环Prins环化反应顺利发生, 以45%的收率得到顺式[5-7]并环化合物109. 化合物105在三光气和DBACO的条件下消除叔醇, 接着脱除TBDPS保护, 随后利用Carreira钴催化剂(106)催化的Mukaiyama水合反应引入C(10)的叔醇, 同样以7步完成了ophiopogonol A的合成.
9 杨震课题组不对称合成buforspirostenin A
2022年, 杨震课题组[38]也报道了一条bufospiro- stenin A的简洁合成路线(Scheme 11). 以tigogenin lac- tone (112)为起始原料, 在(PhSeO)2O/吡啶的条件下将其氧化成双烯酮, 选择性地氢化二取代双键, 通过Mander试剂引入酯基, 最后再发生氧化反应, 以4步32%的产率得到双烯酮酯113. 化合物113在光照的条件下顺利发生山道年重排, 最后经脱羧反应, 以2步38%的总产率得到化合物114. 利用LiAlH(OtBu)3选择性地对114的烯酮进行1,2-还原反应, 并保护为MOM醚, 以2步52%的产率得到化合物115. 化合物115在钴催化剂116和苯硅烷的作用下, 顺利发生可逆的双键异构化, 以56%的产率得到化合物117. 最后采用与李闯创课题组类似的螺环构建策略, 完成了bufospirostenin A的简洁合成.
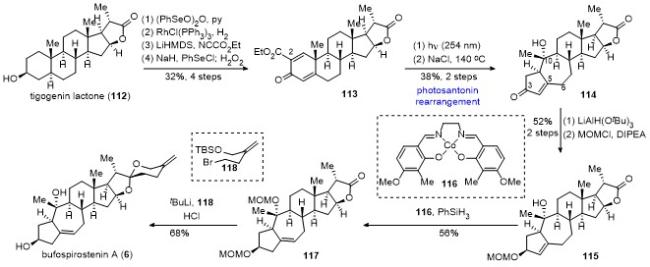
10 李闯创课题组不对称合成phomarol
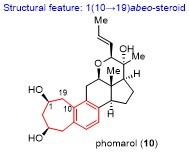
具体合成路线如Scheme 12所示. 以商业可得的化合物119为原料, 经四氟硼酸锂脱除缩醛保护和DIBAL还原, 所得一级羟基用三异丙基硅基(TIPS)保护, 二级羟基用Ac保护, 接着用二氧化硒进行烯丙位氧化, 在C(12)位引入羟基, 最后采用Ley氧化和Luche还原, 调整C(12)位的羟基构型, 以5步22%的总收率得到化合物120. 化合物120的C(12)位羟基先用对甲氧基苄基(PMB)保护, 随后脱除Ac保护并氧化得到烯酮, 其经碘代反应和Stille偶联反应, 以及1,2-加成反应引入C(8)位的高烯丙基, 并脱除呋喃片段上的TBS保护, 以5步47%的总收率得到关键前体化合物123. 化合物123在m-CPBA的条件下发生Achmatowicz氧化重排, 随后原位加入三氟乙酸, 所期望的type I [5+2]环加成反应顺利发生, 以“一锅法”60%的收率得到[5+2]环加成产物124.
利用二氯亚砜和吡啶消除中间体124的C(8)位羟基, 然后“一锅法”对烯酮进行1,2-还原反应, 再经Barton-McCombie脱氧反应, 以3步68%的总收率得到化合物125. 利用TMSOTf选择性打开化合物125的C(10)位的C—O键, 接着发生芳构化反应, 随后加入盐酸脱除TMS保护, 以70%的收率得到化产物126.
化合物126的七元环双键经NBS/H2O处理并加入KOH生成立体选择性的环氧, 用LiAH4区域选择性地开环氧, 保护为MOM醚, 脱除侧链的TIPS保护, 氧化为醛, 接着在叔丁醇钾和氧气的作用下发生去甲酰化反应, 以6步39%的总收率得到酮化合物127. 采用碘代物(R)-35衍生的锂试剂, 对化合物127进行立体选择性的1,2-加成反应得到双烯丙醇, 其在双(三甲基硅烷基)氨基钾(KHMDS)和Ts-imidazole (Ts-Im)的作用下生成环氧, 以2步66%的收率得到化合物128. 利用2,3-二 氯-5,6-二氰基-1,4-苯醌(DDQ)脱除环氧化合物128的C(12)位的PMB保护, 随后在盐酸的作用下顺利关上四氢吡喃环, 完成了phomarol的首次不对称全合成.
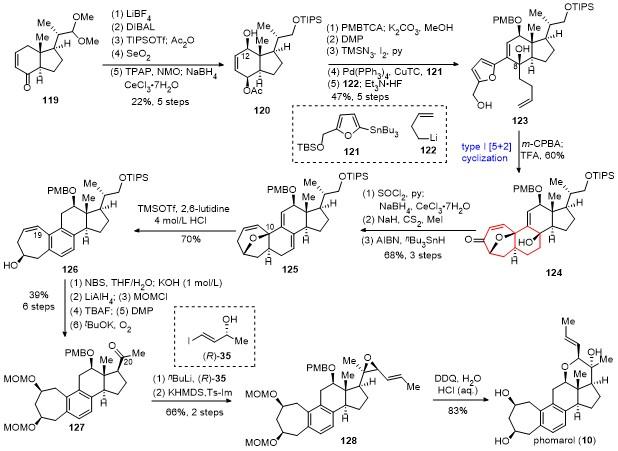
11 桂敬汉课题组不对称全合成phomarol
2023年, 基于生源假说策略, 桂敬汉课题组[41]报道了第二例phomarol全合成. 具体合成路线如Scheme 13所示. 以商业可得的谷内酯(sitolactone, 129)为原料, 经内酯开环和氧化, 所得的酮羰基进行缩酮保护, 随后通过钯催化的脱羧消除, 引入C(8)位的乙烯基, 以3步27%的总收率得到化合物130. 随后利用Schönecker- Baran碳氢键氧化策略, 引入C(12)位的羟基, 并通过van Leusen反应在C(17)位引入氰基, 以3步58%的总收率得到化合物131. 化合物131的氰基和甲基格氏试剂反应得到甲基酮, 接着C(12)位的羟基用TES保护, 然后利用化合物(S)-35衍生的锂试剂对甲基酮进行选择性的1,2-加成反应, 得到关键中间体132. 在KHMDS和Ts-Im的作用下, 化合物132的C(12)位羟基TES保护基首先迁移到C(20)位的羟基上, 接着C(24)位羟基发生磺酸酯化反应, C(12)位的氧负离子与C(22)位的双键发生SN2'关环, C(24)位的磺酸酯基团离去, 加入TBAF原位脱除C(20)位的TES保护, 立体选择性地构建了四氢吡喃环, 最后脱除缩酮保护, 以2步34%的收率得到化合物133. 需要指出的是, C(24)位羟基的手性和离去基团的选择对四氢吡喃环的手性中心控制至关重要. 化合物133的C(9)位的酮羰基在KHMDS和PhNTf2的存在下, 生成烯基三氟甲磺酸酯, 随后与硼试剂139发生Suzuki偶联反应, 以2步63%的收率得到化合物134. 硼试剂139可以通过已知手性醛化合物135经不对称加成反应、TBS保护反应、RCM关环和钯催化的硼化反应得到. 化合物134在加热条件下, 发生6π电环化反应, 接着氧化芳环化, 并原位加入TBAF脱除硅基保护, 完成了phomarol的合成.
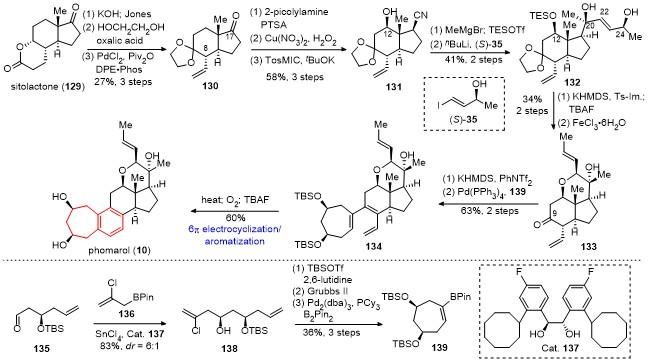
对于abeo-甾体phomarol分子, 李闯创课题组采用type I [5+2]环加成反应、酸促进的芳构化反应和合成后期SN2开环氧构建吡喃环为关键策略, 首次实现了phomarol的不对称全合成; 而桂敬汉课题组通过合成早期的仿生SN2'反应构建吡喃环, 接着6π电环化和芳构化反应为关键策略, 实现了第二例phomarol的不对称全合成.
12 李闯创课题组不对称全合成bufogar- garizins A和B
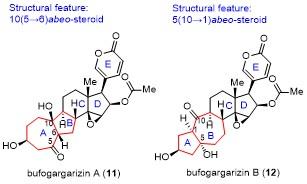
具体合成路线如Scheme 14所示. 以易得的醛酮化合物140(从商业可得的古内酯130经3步转化得到)为起始原料, 经格氏试剂141对醛基的加成反应所得的醇用TES保护, 通过van Leusen反应在C(9)位引入氰基, DIBAL还原氰基为醛, 并在碱性条件下调整C(9)位的手性中心, 以4步45%的总收率得到化合物142.
化合物142与Ohira-Bestmann试剂86发生Seyferth- Gilbert增碳反应引入端炔, 选择性脱除TES保护并氧化, 接着在TMSOTf条件下, 制备成烯醇硅醚143 (Z/E=2.5/1), 其在CpRu(CH3CN)3PF6的催化下, 顺利发生乙烯基醚环丙烷(VECP)参与的[5+2]环加成反应, 以3步56%的总收率得到四环骨架化合物144.
化合物144经Mukaiyama水合反应引入C(10)位的叔醇、DIBAL还原C(5)位酮羰基、脱除TBS保护、DMP氧化二醇为二酮及双Saugusa氧化引入A环和D环的烯酮, 以4步40%的总收率得到化合物145. 化合物145在硅胶和N,N-二异丙基乙胺(DIPEA)条件下, C(15)- C(16)位双键迁移到C(14)-C(15)位, 碱性条件下对A环的烯酮进行环氧化反应, 接着Mukaiyama水合反应引入C(14)位的叔醇并用TMS保护, 以3步46%的总收率得到化合物146. 化合物146在KHMDS和Comins试剂条件下转化为烯基三氟甲磺酸酯, 接着采用SmI2打开环氧, 所得的醇用MOM保护, DIBAL还原酮羰基, 所得的醇用TMS保护, 以3步52%的总收率得到化合物147.
化合物147与吡喃酮148发生Suzuki偶联, 所得产物在m-CPBA的条件下, 立体选择性地对D环的双键进行环氧化, 随后加入硼烷, 原位发生烯基还原-开环氧反应, 以2步64%收率得到化合物149. 对化合物146的C(16)位的羟基进行DMP氧化, 随后在DBU的条件下发生β-羟基消除, 同时发生双键迁移异构为吡喃酮, 而C(17)位手性中心用LHMDS作碱进行动力学异构化, 以2步68%的收率得到化合物150. 化合物150经Corey- Bakshi-Shibata (CBS)还原并用Ac保护所得的醇, 利用NBS/H2O条件下立体选择性地环氧化, 脱除TMS保护, DMP氧化C(5)位的羟基为酮, 脱MOM保护, 最终以4步28%的产率完成了bufogargarizin A的首次不对称全合成.
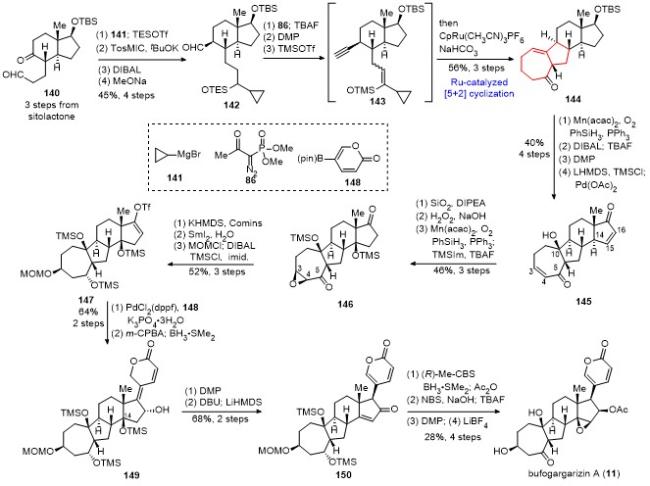
完成了bufogargarizin A的全合成后, 受bufogar- garizins A和B的生源假说[43]启发, 从化合物144出发, 利用合成bufogargarizins A中类似的转化(144→146), 以5步30%的总收率得到化合物151 (Scheme 15). 经过大量的条件筛选, 发现在DBU作碱, 四氢呋喃作溶剂, 以及回流的条件下, 仿生的逆aldol/跨环aldol串联反应顺利发生, 实现了环系的重构, 以73%的收率得到单一构型的产物153. 化合物153经过DIBAL还原、脱TBS保护和DMP氧化, 以2步51%的收率得到化合物154. 化合物154经过bufogargarizin A相似的合成途径, 最终实现了bufogargarizins B的首次不对称全合成.
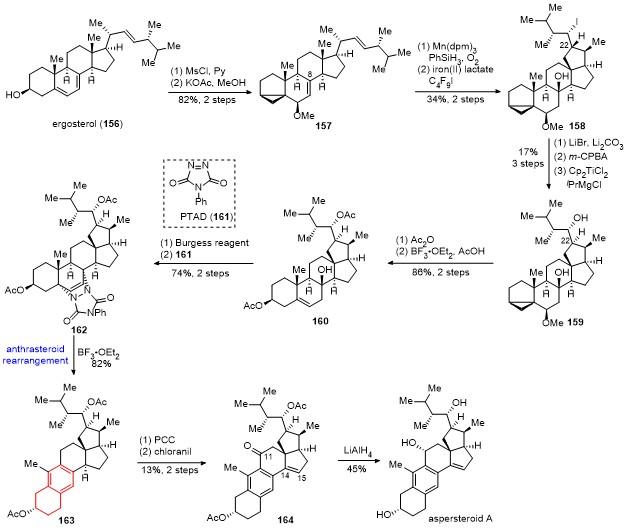
13 桂敬汉课题组不对称合成aspersteroid A
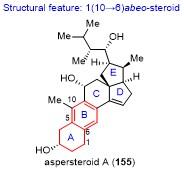
具体合成路线如Scheme 16所示. 从商业可得的麦角固醇156经2步转化得到157, 其经Mukaiyama水合反应在C(8)位引入过氧叔醇, 接着发生Fe(II)介导的自由基接力环化反应, 以2步34%的收率得到碘代物158. 化合物158经消除反应、环氧化反应和Ti(III)介导的立体选择性的还原开环氧反应, 以3步17%的总收率得到C(22)位具有正确构型的化合物159. 化合物159的二级羟基用Ac保护, 接着打开三元环, 以2步86%的收率得到化合物160. 采用Burgess试剂对化合物160的三级羟基进行区域选择性的消除, 所得双烯体与化合物161发生[4+2]环加成反应, 以2步74%的收率得到化合物162. 在三氟化硼乙醚条件下, 化合物162顺利发生anthrasteroid重排反应[46], 以82%的收率得到苯环化合物163. 采用氯铬酸吡啶鎓盐(PCC)对化合物163的C(11)位进行选择性的氧化, 接着采用chloranil试剂引入C(14)-C(15)位的双键, 以2步13%的收率得到化合物164. 化合物164经氢化铝锂还原, 以45%的收率得到天然产物aspersteroid A.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
桂敬汉课题组以15步反应完成了abeo-甾体aspersteroid A的合成. 合成亮点主要包括: (1) Fe(II)介导的过氧均裂引发的自由基接力环化反应, 基于仿生串联重排反应高效构建E环体系; (2) Ti(III)介导的立体选择性的还原开环氧反应构建C(22)位手性中心; (3)三氟化硼乙醚促进的anthrasteroid重排反应构建苯环.
14 结论与展望
总结了自2018年起报道的abeo-甾体全合成研究进展. 在此期间(2018年至2025年5月), 有5个研究团队(李闯创、桂敬汉、Heretsch、马志强和杨震)共报道了24个abeo-甾体的全合成. 这24个abeo-甾体与经典甾体的[6-6-6-5]四环母核结构已大不相同. 比如cyclo- citrinol及其家族分子具有[7-7-6-5]四环体系, 且AB环系为[7-7]桥环体系(双环[4.4.1]桥环体系); swinhoei- sterols A~C具有[6-6-5-7]四环并环体系; dankasterones A和B具有[6-6-5-6]四环体系, 且C、D环系为[5-6]螺环体系; periconiastone A具有[6-6-5-6]四环基本骨架, 但在B环上又增加了一个双环[2.2.2]桥环体系; buforspirostenin A和ophiopogonol A具有[5-7-6-5-5-6]六环并环体系, 其中E、F环系为缩酮[5-6]螺环体系; phomarol具有[7-6-6-5-6]五环并环体系, 其中B环是一个独特的苯环结构; bufogargarizin A具有[7-5-6-5]四环并环体系, 而bufogargarizin B具有[5-7-6-5]四环并环体系.
abeo-甾体结构复杂多变, 具有多个手性中心, 其全合成具有很大的挑战性. 近年来, 通过创新的合成策略和方法在abeo-甾体全合成上取得了巨大突破[47]. 这些创新的策略和方法主要有环加成反应、仿生重排反应、自由基环化反应、偶联反应和6π电环化反应等.
由于abeo-甾体在自然界中分离提取的产率极低, 其后续生物活性研究因此受到限制, 而这些创新的合成路线, 为获得更充足的abeo-甾体及其类似物提供了替代方案. 通过天然产物合成的不断创新, 将不仅有助于理解abeo-甾体的结构及其生源转化, 也为后续的药物研发提供了物质保障.
(Zhao, C.)