

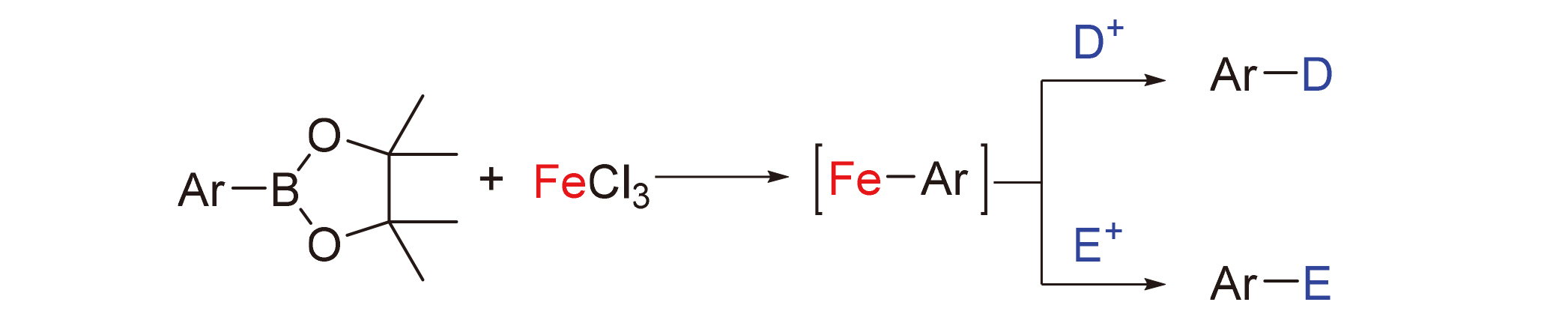
1 Introduction
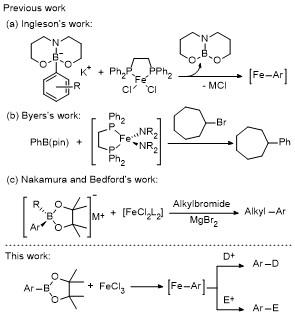
Fe-catalyzed/mediated organic transformations have received increasing interest because Fe-complexes have the advantages of low cost, non-toxicity and good environmental sustainability.[1-6] In the past decades, approaches for C—Fe bond construction from C—B bond[7] have been well-developed. For example, Ingleson et al.[8] reported a Fe(II)-mediated transmetallation with organoboronate activated by 3,3'-azanediyldipropan-1-ol, giving an isolable organoironic product in good yield (Scheme 1a). Byers [9] reported a strong electron-donating bidentated ligand coordinated Fe(II) complex, which could efficiently cleave C—B bond without any pre-activation process (Scheme 1b). Bedford[10] and Nakamura[11] have independently reported bidentated ligand coordinated Fe(II)-complex catalyzed cross-coupling reactions of organoboronic reagents with alkyl halides, in which C—Fe containing species generated via transmetallation step were speculated as an intermediate (Scheme 1c). With these great achievements, trans- metallation step from C—B to C—Fe has been widely accepted. However, the factors which influence the trans- metallation process are not well-integrated, including the ligand effect, spin-state and oxidation state of Fe cation, necessity for pre-activation of C—B bond with nucleophiles and the assistance of other metal halides.[8-9,11-17] Also, it is challenging to characterize the active iron intermediate in a live system.[18-20] Therefore, it is necessary to further develop a concise and efficient method for Fe-me- diated C—B bond cleavage, supplying stoichiometric information for C—Fe bond construction and reactivity. In this paper, we report a concise Fe(III)-mediated C—B cleavage reaction. The transmetallation reaction can be accomplished in the absence of ligand, without valance alternation of Fe(III). C—Fe bond-containing intermediate can be successfully isotope-labelled and trapped by strong electrophiles. Pinacol substituent of organoboronate reagents was found crucial for accelerating deboronation process. Lability and thermal instability of C—Fe(III) bond on the aryl group has also been observed.
2 Results and discussion
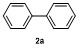
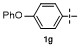
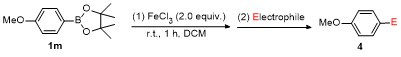
At the beginning of our work, pinacol 4-biphenylboro- nate 1a was employed as the substrate. As shown in Table 1, using 2.0 equiv. of anhydrous FeCl3 as the deboronation reagent, 1a could smoothly undergo C—B cleavage at 100 ℃ in 5 min. After quenching the reaction mixture with H2O, biphenyl can be isolated in 84% yield (Entry 1). When AlCl3 was used instead of FeCl3, the reaction proceeded vigorously: substrate 1a was completely decomposed within 5 min, and product 2a was detected in 15% yield via NMR analysis (Entry 2). In contrast, when CuCl2 was used instead, no reaction occurred even in prolonged reaction time (Entry 3). In Entries 4 and 5, anion effect of Fe(III) salt was tested, in which Fe2(SO4)3 and Fe(acac)3 were found quite ineffective to C—B cleavage. In Entry 4, product 2a was formed in only 7% isolated yield. In Entry 5, 2a could not be detected. In Entries 6, 7 and 8, Fe(II)Cl2, Fe(OAc)2 and Fe2O3 were investigated, and no deboronated product 2a was formed after quenched. Solvent effect for this reaction was further investigated. In Entries 9 and 10, the reaction could not proceed when tetrahydrofuran (THF) or N,N-dimethylformamide (DMF) was used as the solvent. In Entry 11, with CHCl3 as the solvent, the reaction could proceed slowly and the yield of 2a reached 61% after 90 min. In Entry 12, the stoichiometry of Fe(III)Cl3 was compromised, and the yield of 2a was found decreased to 18%. In Entry 13, when the reaction was performed at room temperature, 2a could also be obtained in good yield after 30 min. The effect of pinacol substituent was also investigated, as pinacolone was detected by NMR and GC analysis in Entry 13. In Entry 14, with 4-biphen- ylboronic acid as the substrate, the deboronation reaction resulted in a complicated mixture, and the isolated yield of 2a was greatly decreased. In Entry 15, replacing the pinacol substituent with i-PrO resulted in highly reluctant C—B bond cleavage, with only a detectable amount of biphenyl formed, implying the significant accelerating effect of pinacol substituent. Thus, the optimal conditions for the deboronation reaction are 2.0 equiv. of FeCl3 as the reagent and CH2Cl2 as the solvent at 25 ℃, with pinacolboronate as the substrate.
Table 1 Condition optimizations for C—B bond cleavage |
| Entry | [M] (equiv.) | Solvent | T/℃ | t/min | Yielda/% |
|---|---|---|---|---|---|
| 1 | FeCl3 (2.0) | CH2Cl2 | 100 | 5 | 84 |
| 2 | AlCl3 (2.0) | CH2Cl2 | 100 | 5 | 15 |
| 3 | CuCl2 (2.0) | CH2Cl2 | 100 | 30 | N.D.b |
| 4 | Fe2(SO4)3 (1.0) | CH2Cl2 | 100 | 180 | 7 |
| 5 | Fe(acac)3 (2.0) | CH2Cl2 | 100 | 60 | N.D. |
| 6 | Fe(OAc)2 (2.0) | CH2Cl2 | 100 | 60 | N.D. |
| 7 | FeCl2 (2.0) | CH2Cl2 | 100 | 60 | N.D. |
| 8 | Fe2O3 (2.0) | CH2Cl2 | 100 | 60 | N.D. |
| 9 | FeCl3 (2.0) | THF | 100 | 120 | N.D. |
| 10 | FeCl3 (2.0) | DMF | 100 | 120 | N.D. |
| 11 | FeCl3 (2.0) | CHCl3 | 100 | 90 | 61 |
| 12 | FeCl3 (1.0) | CH2Cl2 | 100 | 90 | 18 |
| 13 | FeCl3 (2.0) | CH2Cl2 | 25 | 30 | 87 |
| 14c | FeCl3 (2.0) | CH2Cl2 | 25 | 60 | 49 |
| 15d | FeCl3 (2.0) | CH2Cl2 | 25 | 60 | <5 |
a Isolated yield. b N.D.=not detected. c 4-Biphenylboronic acid was used as the substrates. d Diisopropyl[1'-biphenyl]-4-ylboronate was used as the subs- trates. |
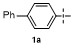
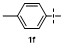
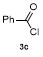
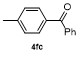
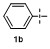
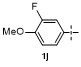
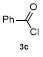
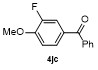
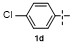
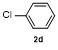
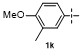
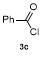
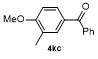
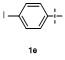
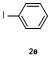
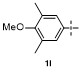
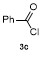
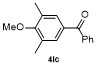
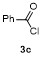
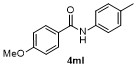
Under the optimized condition, the substrate scope of aryl boronates was investigated (Table 2). 2a was isolated in 87% yield after deboronation at 25 ℃ (Entry 1). When pinacol phenylboronate 1b was employed, deboronation could proceed at elevated 60 ℃, and benzene was detected in 85% yield in 1 h (GC analysis) after the reaction was quenched with H2O (Entry 2). With 1c as the substrate, C—B cleavage occurred at 25 ℃ and naphthalene was formed in 64% yield based on 1H NMR (Entry 3). With electron deficient substrates 1d and 1e, the reactions were performed at 100 ℃ for 3 and 4 h, and 2d and 2e were formed in 58% and 52% yields (GC analysis), respectively (Entries 4 and 5). In the following Entries, acyl chloride 3c instead of H2O was employed to facilitate the isolation and clarify the regioselectivity as well as isolated yields of the products. In Entry 6, deboronated 1f reacted with benzoyl chloride and 4fc was isolated in 51% yield. In Entry 7, with phenoxy group as the substituent, 4gc could be obtained in 68% isolated yield. With electron-withdrawing groups introduced, acylation could not proceed. However, polysubstituted phenylboronates was found reactive. In Entry 8, fluoro-substituent was introduced to p-methoxy phenylboronate, and 4jc was formed exclusively in 63% yield. In Entries 9 and 10, the introduction of one or two methyl groups to p-methoxy phenylboronate could not influence the regioselectivity, with 4kc and 4lc obtained in 66% and 86% isolated yields, respectively.
Table 2 Scope of arylboronatesa |
| Entry | Aryl | Electrophile | Product | Yieldb/% of 2 | Entry | Aryl | Electrophile | Product | Yieldb/% of 4 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | | H2O | | 87 | 6 | | | | 51 |
| 2c | | H2O | | 85d | 7 | | | | 68 |
| 3 | | H2O | | 64e | 8 | | | | 63 |
| 4f | | H2O | | 58d | 9 | | | | 66 |
| 5g | | H2O | | 52d | 10 | | | | 86 |
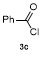
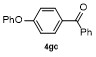
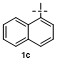
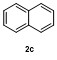
a Condition: 0.2 mmol of 1, at 25 ℃ for 1 h in 2 mL of CH2Cl2, followed 2.0 equiv. of electrophile (except H2O), stirring for 4 h at 25 ℃. b Isolated yields. c At 60 ℃ for 1 h. d GC yield using tetradecane as internal standard. e NMR yield using mesitylene as internal standard. f 3 h at 100 ℃. g 4 h, at 100 ℃. |
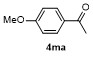
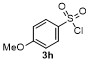
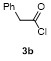
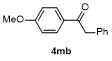
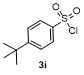
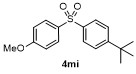
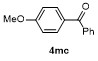
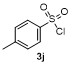
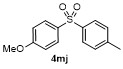
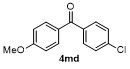
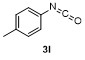
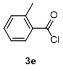
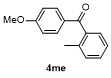
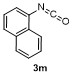
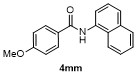
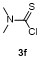
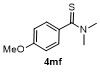
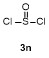
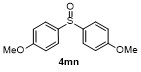
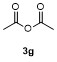
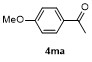
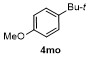
The cross-coupling reactions of arylboronate with acyl chloride were usually achieved via Pd-catalysis.[21-23] Such Pd-catalyzed coupling reactions, which deliver good to excellent yields, are widely taken as model reactions for testing catalytic reactivity of Pd-complexes. To the best of our knowledge, Fe-mediated coupling reaction of arylboronic ester with acyl chloride is rare in the literature. Based on the research in Table 2, electron-rich phenylironic intermediate was found more suitable for investigating C—Fe reactivity. Thus, the scope of electrophiles was further investigated, with p-methoxyl phenyl pinacol boronate 1m as the substrate (Table 3). In Entry 1, 3a could be successfully incorporated to give 4ma in 68% yield. 2-Phenylacetyl chloride 3b gave a compatible result, and 4mb was isolated in 65% yield (Entry 2). Aroyl chlorides were proved more active in this reaction. Phenyl group with p-Cl- and o-Me-substituents were all tolerant to this condition, with 4mc, 4md and 4me obtained in 81%, 81% and 79% isolated yields, respectively (Entries 3~5). When less active 3f was employed as the substrate, coupling product 4mf was obtained in only 15% isolated yield, with most of anisole recovered, proving the weak nucleophilicity of organoiron intermediate (Entry 6). Acetic anhydride 3g was proved quite reactive to 1m, affording 4ma in 85% yield (Entry 7). In case sulfonyl chloride 3h was employed, 4mh was obtained in 60% yield (Entry 8). In contrast, with t-butyl and methyl groups as the substituent, the yields of coupling products 4mi and 4mj decreased to 23% and 21% yields, with anisole remained in the mixture (Entries 9 and 10). When isocyanates 3l and 3m were used as substrates (Entries 11 and 12), though the coupling reaction proceeded well, products 4ml and 4mm tend to in situ decompose to aryl amines, thereby resulting in moderate to low yields (51% and 30%). Thionyl chloride 3n was also proved active to organoiron intermediate, and 4mn was formed in 62% isolated yield (Entry 13). When alkyl halides were employed, MeI, allylic chloride and cyclohexyl bromide were found all inert in the reactions, except 3o afforded 4mo in 55% yield (Entry 14).
Table 3 Scope of electrophilesa |
| Entry | E+ | Product | Yieldb/% | Entry | E+ | Product | Yieldb/% |
|---|---|---|---|---|---|---|---|
| 1 | | | 68 | 8 | | | 60 |
| 2 | | | 65 | 9 | | | 23 |
| 3 | | | 81 | 10 | | | 21 |
| 4 | | | 81 | 11 | | | 51 |
| 5 | | | 79 | 12 | | | 30 |
| 6 | | | 15 | 13 | | | 62 |
| 7 | | | 85 | 14 | | | 55 |
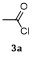
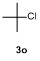
a Condition: 0.2 mmol of 1m, 2.0 equiv. of FeCl3, 2.0 equiv. of electrophiles. b Isolated yields. |
To differentiate the reaction of organoiron intermediated versus classical Friedel-Crafts acylation, the contrast reactions were performed. As shown in Scheme 2, 1b was deboronated with FeCl3 under the best condition in Table 2, followed by allowing to react with benzoyl chloride. After 8 h, hardly detectable benzophenone was formed. In con- trast, under the same condition, with benzene as the substrate, benzophenone was generated in 69% yield (based on NMR analysis). This result proved the reactivity of acylium cation to benzene in Friedel-Crafts acylation is higher than that of organoiron intermediate to acyl chloride.

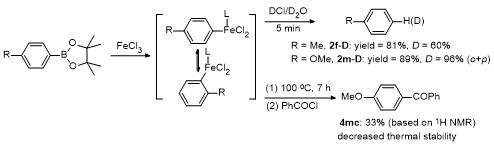
Isotope labelling experiments were performed to confirm the formation of C—Fe(III) bond from C—B bond (Scheme 3). With 1f as the substrate, the protodeboronation intermediate 1f was quenched with DCl (20% in D2O) in 5 min at room temperature. Accordingly, 2f-D was formed in 81% yield with 60% deuteration degree. With more active 1m as the substrate, 2m-D was formed in 89% yield with 96% deuteration degree. These results clearly proved the formation of C-Fe(III) containing intermediate. Surprisingly, the D-labelled sites of 2m-D were found both on ortho- and para-positions of anisole, with a ratio of ortho∶para=68%∶28% based on 1H NMR analysis, indicating a labile regioselectivity on the phenyl ring. The thermal stability of in situ formed organoiron intermediate was also investigated. After keeping heating the organo- iron intermediate for 7 h at 100 ℃, the intermediate was allowed to react with benzoyl chloride, and the yield of coupling product decreased from 81% to only 33%, implying its thermally instability of the intermediate.
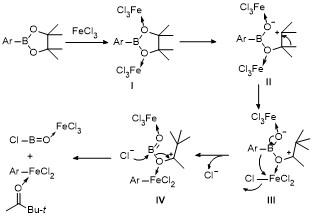
A plausible mechanism was shown in Scheme 4. In the first step, 2.0 equiv. of FeCl3 coordinated to oxygen atoms of aryl boronate as a Lewis acid, inducing a C—O bond of pinacol substituent cleavage to give a carbocationic species II. Intermediate II underwent pinacol-type rearrangement to give a pinacolone skeleton-containing intermediate III. Thereafter, the anionic oxygen generated stable B=O bond, which promoted aryl group to undergo nucleophilic attack to Fe(III) center, giving intermediate IV containing Ar—Fe bond. In this step, C—B cleavage is dependent on electronic property of aryl group. A further nucleophilic attack of Cl- to boron center in IV led to the formation of pinacolone. According to this mechanism, the mixture of deboronation reaction was characterized, and pinacolone was found to be generated quantitively according to NMR analysis, which is consistent with the proposed mechanism.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 Conclusions
A concise and efficient method for FeCl3-mediated C—B cleavage was accomplished. A series of electrophilic reagents have been proved reactive to this organoiron intermediate. The in situ generated organoiron intermediate can be labelled with deuterium. The labile character of C—Fe bond on aryl group has been observed. The C—B cleavage process is found to be accelerated by pinacol substituent. Further formation mechanism and reactivity investigation of this organoiron intermediate will be reported in due course.
4 Experimental section
4.1 General information
Unless otherwise stated, all reactions were carried out under N2 atmosphere using Schlenk technique and glove box. Solvents were dried by sodium hydride and distilled under nitrogen. All reactions were monitored by thin layer chromatography, gas chromatography or 1H NMR. Purification of reaction products was carried out by flash chromatography on silica gel. Chemical yields refer to pure isolated substances. 1H NMR and 13C NMR spectra were recorded on Bruker 400 MHz spectrometers (400 MHz for 1H NMR and 101 MHz for 13C NMR). Chemical shifts of 1H and 13C signals were given in δ relative to the solvents residual 1H-signal (CHCl3, δ 7.26, DMSO, δ 2.50), or Me4Si (δ 0.0). CDCl3 resonance in the 13C spectrum is δ 77.5 and DMSO resonance in the 13C spectrum is δ 39.5. High-resolution mass spectral analysis (HRMS) data were measured on a Bruker ApexII mass spectrometer by means of the ESI technique.
4.2 General procedure for deboronation and the synthesis of compounds 4
Anhydrous iron(III) chloride (0.4 mmol, 64.9 mg) was transferred from glove box to Schlenk tube equipped with a magnetic bar. Iron(III) chloride was further dried with hot air gun under reduced pressure. After cooling down, Schlenk tube was refilled with nitrogen and evacuated. After performing the procedure for three times, dichloromethane (2 mL) was added to the Schlenk tube, and arylboronic acid pinacol ester (0.2 mmol) was added into the Schlenk tube. The Schlenk tube was then sealed tightly, and the mixture was stirred at room temperature for 1 h. Thin-layer chromatography (TLC) analysis could prove the complete consumption of arylboronates and generation of organoiron intermediates. For the synthesis of compounds 4, 0.4 mmol of electrophile was added to the above mixture and kept stirring. After monitored by TLC analysis, the solid residue was filtered and the solution was evacuated under reduced pressure. The resulted residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate or dichloromethane as an eluent) to afford the desired compounds 4.
Phenyl(p-tolyl)methanone (4fc):[24] White solid, 20.0 mg, yield 51%. m.p. 40 ℃ (lit.[25] 43.1~44.3 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.78 (d, J=7.2 Hz, 2H), 7.72 (d, J=8.0 Hz, 2H), 7.57 (t, J=7.2 Hz, 1H), 7.47 (t, J=7.6 Hz, 2H), 7.29~7.27 (m, 2H), 2.44 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 197.0, 143.7, 138.4, 135.4, 132.6, 130.8, 130.4, 129.5, 128.7, 22.1.
(4-Phenoxyphenyl)(phenyl)methanone (4gc):[26] White solid, 37.3 mg, isolated yield 68%. m.p. 76 ℃ (lit.[26] 68~69 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.83~7.77 (m, 4H), 7.57 (t, J=7.2 Hz, 1H), 7.47 (t, J=7.6 Hz, 2H), 7.40 (t, J=7.6 Hz, 2H), 7.20 (t, J=7.2 Hz, 1H), 7.09 (d, J=8.0 Hz, 2H), 7.03 (d, J=8.8 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ: 196.0, 162.1, 155.9, 138.4, 132.9, 132.6, 132.3, 130.5, 130.3, 128.7, 125.1, 120.6, 117.6.
(3-Fluoro-4-methoxyphenyl)(phenyl)methanone(4jc):[27] White solid, 29.0 mg, isolated yield 63%. m.p. 104~106 ℃ (lit.[27] 67~68 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.75 (d, J=7.2 Hz, 2H), 7.64~7.58 (m, 3H), 7.48 (t, J=7.6 Hz, 2H), 7.00 (t, J=8.4 Hz, 1H), 3.97 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 195.0, 152.2 (d, 1JC-F=249.5 Hz), 152.0 (d, 2JC-F=11.1 Hz), 138.0, 132.7, 130.8 (d, 3JC-F=5.1 Hz), 130.2, 128.8, 128.2 (d, 4JC-F=4.0 Hz), 118.2 (d, 2JC-F=19.2 Hz), 112.6, 56.8.
(4-Methoxy-3-methylphenyl)(phenyl)methanone(4kc):[28] Colorless oil, 29.9 mg, isolated yield 66%. 1H NMR (400 MHz, CDCl3) δ: 7.76~7.74 (m, 2H), 7.69~7.66 (m, 2H), 7.57~7.53 (m, 1H), 7.48~7.45 (m, 2H), 6.86 (d, J=8.4 Hz, 1H), 3.90 (s, 3H), 2.26 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 196.3, 162.0, 138.9, 133.2, 132.2, 131.1, 130.2, 130.1, 128.6, 127.2, 109.4, 56.0, 16.7.
(4-Methoxy-3,5-dimethylphenyl)(phenyl)methanone (4lc):[29] White solid, 41.3 mg, isolated yield 86%. m.p. 46 ℃ (lit.[30] 49 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.77 (d, J=7.2 Hz, 2H), 7.57 (t, J=7.6 Hz, 1H), 7.49~7.45 (m, 4H), 3.78 (s, 3H), 2.33 (s, 6H); 13C NMR (101 MHz, CDCl3) δ: 196.8, 161.2, 138.4, 133.5, 132.5, 131.6, 131.4, 130.3, 128.6, 60.12, 16.7.
1-(4-Methoxyphenyl)-2-phenylethan-1-one (4mb):[33] White solid, 29.4 mg, isolated yield 65%. m.p. 78~80 ℃ (lit.[34] 76~78 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.99 (d, J=8.8 Hz, 2H), 7.33~7.21 (m, 5H), 6.91 (d, J=8.8 Hz, 2H), 4.22 (s, 2H), 3.84 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 196.7, 163.9, 135.4, 131.4, 130.0, 129.8, 129.1, 127.2, 114.2, 55.9, 45.7.
(4-Methoxyphenyl)(phenyl)methanone (4mc):[31] White solid, 34.4 mg, isolated yield 81%. m.p. 68 ℃ (lit.[35] 65.6~66.6 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.82 (d, J=8.8 Hz, 2H), 7.75 (d, J=6.8 Hz, 2H), 7.55 (t, J=7.2 Hz, 1H), 7.46 (t, J=7.6 Hz, 2H), 6.95 (d, J=8.8 Hz, 2H), 3.87 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 195.9, 163.6, 138.7, 133.0, 132.3, 130.5, 130.1, 128.6, 114.0, 55.9.
(4-Chlorophenyl)(4-methoxyphenyl)methanone (4md):[36] White solid, 39.9 mg, isolated yield 81%. m.p. 134~136 ℃ (lit.[37] 128~130 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.79 (d, J=8.4 Hz, 2H), 7.70 (d, J=8.0 Hz, 2H), 7.44 (d, J=8.0 Hz, 2H), 6.96 (d, J=8.4 Hz, 2H), 3.88 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 194.6, 163.8, 138.6, 136.9, 132.9, 131.6, 130.1, 128.9, 114.1, 55.9.
(4-Methoxyphenyl)(2-tolyl)methanone (4me):[38] Yellow oil, 35.7 mg, isolated yield 79%. 1H NMR (400 MHz, CDCl3) δ: 7.79 (d, J=8.8 Hz, 2H), 7.38~7.34 (m, 1H), 7.29~7.21 (m, 3H), 6.92 (d, J=8.8 Hz, 2H), 3.86 (s, 3H), 2.30 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 197.8, 164.1, 139.6, 136.5, 132.9, 131.2, 130.9, 130.2, 128.3, 125.6, 114.1, 55.9, 20.2.
4-Methoxy-N,N-dimethylbenzothioamide (4mf):[39] Yellow solid, 5.9 mg, isolated yield 15%. m.p. 72~74 ℃ (lit.[40] 68~69 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.30 (d, J=8.8 Hz, 2H), 6.86 (d, J=8.8 Hz, 2H), 3.81 (s, 3H), 3.58 (s, 3H), 3.21 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 201.7, 160.4, 136.2, 128.3, 113.9, 55.8, 44.7, 44.0.
1-(t-Butyl)-4-((4-methoxyphenyl)sulfonyl)benzene(4mi):[41] White solid, 14.0 mg, isolated yield 23%. m.p. 158 ℃ (lit.[41] 134.7 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.88 (d, J=9.2 Hz, 2H), 7.83 (d, J=8.4 Hz, 2H), 7.48 (d, J=8.8 Hz, 2H), 6.96 (d, J=9.2 Hz, 2H), 3.83 (s, 3H), 1.30 (s, 9H); 13C NMR (101 MHz, CDCl3) δ: 163.7, 157.1, 139.8, 134.0, 130.3, 127.6, 126.7, 114.9, 56.1, 35.6, 31.5.
1-Methoxy-4-tosylbenzene (4mj):[41] White solid, 11.0 mg, isolated yield 21%. m.p. 104~106 ℃ (lit.[41] 105.5 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.86 (d, J=8.8 Hz, 2H) 7.79 (d, J=8.0 Hz, 2H), 7.27 (d, J=7.6 Hz, 2H), 6.95 (d, J=8.8 Hz, 2H), 3.83 (s, 3H), 2.38 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 163.6, 144.2, 139.8, 133.9, 130.3, 130.1, 127.8, 114.9, 56.1, 22.0.
4-Methoxy-N-(p-tolyl)benzamide (4ml):[42] White solid, 24.6 mg, isolated yield 51%. m.p. 164 ℃ (lit.[43] 155~157 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.83 (d, J=8.8 Hz, 2H), 7.79 (s, 1H),7.50 (d, J=8.0 Hz, 2H), 7.15 (d, J=8.0 Hz, 2H), 6.95 (d, J=8.4 Hz, 2H), 3.86 (s, 3H), 2.33 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 165.7, 162.8, 136.0, 134.4, 130.0, 129.3, 127.7, 120.7, 114.4, 55.9, 21.4.
4-Methoxy-N-(naphthalen-1-yl)benzamide (4mm):[44] White solid, 16.6 mg, isolated yield 30%. m.p. 218~220 ℃ (lit.[45] 205~206 ℃); 1H NMR (400 MHz, DMSO) δ: 10.30 (s, 1H), 8.09 (d, J=8.8 Hz, 2H), 7.99~7.96 (m, 2H), 7.86 (d, J=6.8 Hz, 1H), 7.60~7.53 (m, 4H), 7.10 (d, J=8.8 Hz, 2H), 3.86 (s, 3H); 13C NMR (101 MHz, DMSO) δ: 165.6, 162.0, 134.1, 133.8, 129.7, 129.3, 128.1, 126.5, 126.2, 126.0, 125.9, 125.6, 124.0, 123.4, 113.7, 55.5.
1-(t-Butyl)-4-methoxybenzene (4mo):[48] Colorless oil, 18.1 mg, isolated yield 55%. 1H NMR (400 MHz, CDCl3) δ: 7.31 (d, J=8.8 Hz, 2H), 6.85 (d, J=8.8 Hz, 2H), 3.79 (s, 3H), 1.30 (s, 9H); 13C NMR (101 MHz, CDCl3) δ:157.8, 143.8, 126.7, 113.8, 55.7, 34.5, 32.0.
4.3 Contrast reaction with Friedel-Crafts reaction
Under the general procedure for deboronation, 0.2 mmol of benzene and phenylboronic acid pinacol ester 1b were treated with FeCl3 at 60 ℃ for 1 h, respectively. After 1b was consumed completely based on TLC analysis, 0.4 mmol of benzoyl chloride was charged into the two reactors and the reactions were left stirring for another 8 h at room temperature. Afterwards, the reaction mixture was evaporated under reduced pressure and was filtered through silica gel to remove the insoluble solid. Mesitylene was added to the two obtained samples, followed by rapid evaporation for NMR analysis. Benzophenone was obtained in 69% yield from benzene, while its formation from 1b was hardly detectable.
4.4 Deuterization of 1f and 1m
Under the general procedure for deboronation, anhydrous FeCl3 (1.0 mmol, 162.2 mg) was allowed to react with 0.5 mmol of 1f or 1m for 1 h at 25 ℃. After that, DCl [1.0 equiv., 0.5 mmol, 20% (w) solution in D2O, 75 μL] was added to the mixture and kept stirring for 5 min at 25 ℃. The mixture was allowed to pass through silica gel with dichloromethane as eluent, followed with distillation at 90 ℃ to remove the volatiles. Yields were determined by 1H NMR using mesitylene as internal standard. Deuteration degree was calculated based on 1H NMR.
p-Deuteriotoluene (2f-D):[49] 81% yield. 1H NMR (400 MHz, CDCl3) δ: 7.26~7.23 (m, 2H), 7.18~7.13 (m, 2.4H), 2.35 (s, 3H).
p-Deuterioanisole and o-deuterioanisole (2m-D):[50] 89% yield. 1H NMR (400 MHz, CDCl3) δ: 7.29~7.26 (m, 2H), 6.95~6.88 (m, 2.04H), 3.78 (s, 3H).
Supporting Information 1H NMR and 13C NMR spectra of compounds 4fc, 4gc, 4jc~4lc, 4ma~4mf, 4mh~4mj, 4ml~4mo. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
(Cheng, F.)