
文章编号: A20090409
文献标识码: A
水系锌离子电池研究进展和挑战
 |
张璐, 工学博士, 副教授. 2017年毕业于燕山大学, 曾获得2017年博士后创新人才支持计划, 2018年在美国俄勒冈州立大学从事博士后研究工作, 目前发表文章50余篇, 获授权专利11项, 河北省自然科学奖二等奖1项. 主要研究方向为新型电化学储能体系及器件和储氢材料. |
 |
王文凤, 博士研究生. 2016年起于燕山大学环境与化学工程学院攻读硕士学位, 2018年获得硕博连读资格, 攻读博士学位. 主要研究方向为储氢材料以及水系电池. |
 |
张洪明, 博士研究生. 2015年起于燕山大学环境与化学工程学院攻读硕士学位, 2017年获得硕博连读资格, 攻读博士学位. 主要研究方向为过渡金属催化剂作用下硼氢化合物水解制氢性能的研究. |
 |
韩树民, 博士生导师, 教授. 先后承担并主持完成或承担国家863计划和自然科学基金项目6项、省部级课题7项; 获授权国家发明专利20项, 发表SCI学术论文180篇, 获河北省技术发明奖二等奖2项; 担任Journal of Alloys and Compounds等杂志审稿人, 兼任国际电化学学会会员, 中国电化学委员会委员, 中国材料研究会产业工作委员. 近期的研究工作主要集中在新型稀土功能材料、新型电化学储能系统和器件的研究. |
 |
王利民, 博士生导师, 教授. 2019年入选教育部长江学者特聘教授、新世纪优秀人才支持计划等, 主持国家重点研发计划课题1项、国家自然科学基金5项、中国-意大利国际合作项目2项、科技部国家重点实验室基础研究项目8项、其他省部级项目10余项. 现任《Frontiers in Materials: glass science》副主编. 在Nature, Phys. Rev. Lett., Adv. Mater., J. Phys. Chem. Lett., Acta Mater.等SCI刊物上发表论文169篇, SCI他引超过4000次. 主要研究方向为非晶转变以及玻璃形变过程中的热力学和动力学研究. |
收稿日期:2020-09-04
网络出版日期:2020-11-19
基金资助
国家自然科学基金(51701175)
国家自然科学基金(51971197)
河北省自然科学基金(E2020203081)
河北省自然科学基金(E2019203161)
Research Progress and Challenge of Aqueous Zinc Ion Battery
Received:4 Sept. 2020
Online:19 Nov. 2020
Fund
National Natural Science Foundation of China(51701175)
National Natural Science Foundation of China(51971197)
Natural Science Foundation of Hebei Province(E2020203081)
Natural Science Foundation of Hebei Province(E2019203161)
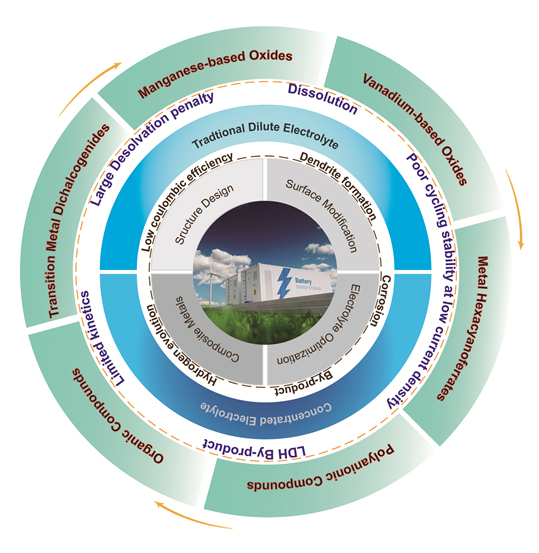
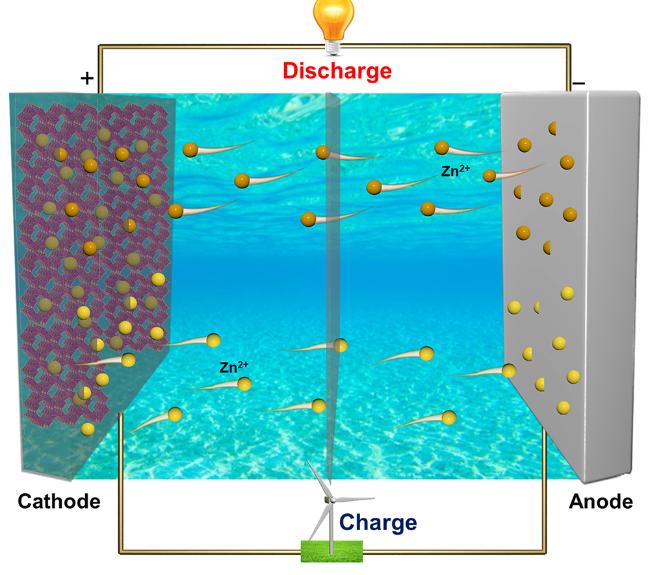
水系锌离子电池(AZIBs)作为一种新兴电池储能技术, 具有安全性高、价格低廉、能量密度高、环境友好、易制造等优点, 在大规模储能等领域具有良好的应用价值和前景, 近年引起了人们的广泛关注且发展迅速. 作者从目前AZIBs存在的科学问题出发, 综述了AZIBs在正负极材料及电解液方面取得的重要进展, 对已开发的多种正极材料的特点及其电化学反应机理进行了分析和总结, 讨论了金属Zn负极面临的挑战和改善策略, 分析了不同水系电解液的特征及其优化方案, 并对这一新兴电池技术面临解决的科学问题和未来实际应用面临的技术挑战进行了总结和展望.
张璐 , 王文凤 , 张洪明 , 韩树民 , 王利民 . 水系锌离子电池研究进展和挑战[J]. 化学学报, 2021 , 79(2) : 158 -175 . DOI: 10.6023/A20090409
Lu Zhang , Wenfeng Wang , Hongming Zhang , Shumin Han , Limin Wang . Research Progress and Challenge of Aqueous Zinc Ion Battery[J]. Acta Chimica Sinica, 2021 , 79(2) : 158 -175 . DOI: 10.6023/A20090409
Aqueous zinc ion batteries (AZIBs) as an emerging battery energy storage technology have attracted widespread attention and developed rapidly in recent years due to the merits of high safety, low price, high energy density, environmental friendliness, easy manufacturing, etc., showing good application value and prospect in large-scale energy storage and other fields. Embarking from the current scientific issues existed in AZIBs, we review the important progress in cathode and anode materials and electrolyte used in AZIBs in this article. Firstly, the characteristics of multiple cathode materials that developed and their electrochemical reaction mechanism are analyzed and summarized. Afterward, the challenge and improving strategy for metal zinc anode are discussed, and the features of different aqueous electrolytes and the optimization solution are analyzed. Finally, the scientific problems to solve and technical challenges for the practical application in the future of this novel battery technology are summarized and prospected.
Key words: aqueous zinc ion battery ; zinc anode ; cathode material ; electrolyte ; energy storage
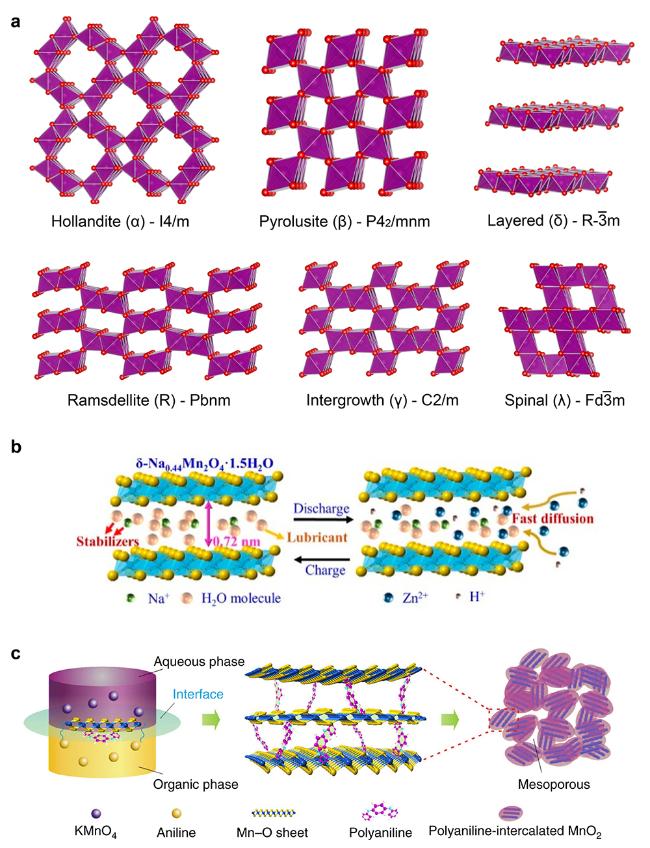
图2 (a) MnO2常见晶体结构图. (b) 层状MnO2中预嵌入Na+提高结构稳定性和离子扩散示意图[18](Copyright 2019, American Chemical Society). (c) 聚苯胺(PANI)插层的MnO2制备示意图[19] (Copyright 2018, Springer Nature)Figure 2 (a) The crystal structure of MnO2. (b) Schematic illustration of the pre-intercalated Na+-MnO2 to increase structural stability and ion diffusion[18] (Copyright 2019, American Chemical Society). (c) Schematic illustration of the preparation of polyaniline (PANI)-intercalated MnO2 [19] (Copyright 2018, Springer Nature) |
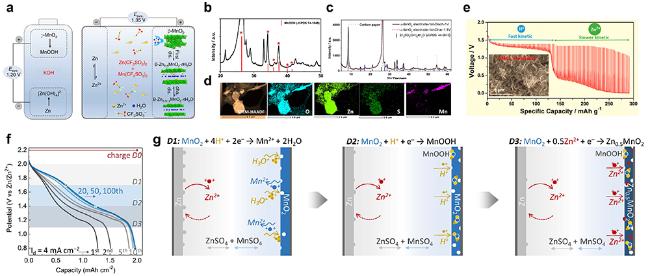
图3 (a) Zn||α-MnO2在3 mol/L Zn(CF3SO3)2+0.1 mol/L Mn(CF3SO3)2电解液中工作机理示意图[24] (Copyright 2017, Springer Nature). (b) α-MnO 2充电后XRD图谱, (c)放电至1.0 V和再充电至1.8 V的XRD图谱和(d)放电至1 V 时STEM-EDS图[20] (Copyright 2016, Springer Nature). (e) Zn||α-MnO2恒电流间歇滴定(GITT)测试图, 内嵌入α-MnO2的SEM形貌图[28] (Copyright 2017, American Chemical Society). ε-MnO 2在1 mol/L ZnSO4+1 mol/L MnSO4电解液中(f) 100周内充放电曲线和(g) 电化学反应机理示意图[31] (Copyright 2019, Wiley-VCH)Figure 3 (a) Schematic illustration of mechanism of Zn||α-MnO2 cell in 3 mol/L Zn(CF3SO3)2+0.1 mol/L Mn(CF3SO3)2 electrolyte[24] (Copyright 2017, Springer Nature). The XRD patterns (b) after fully charged and (c) discharged to 1 V and charged back to 1.8 V of α-MnO 2, and (d) STEM-EDS mappings of α-MnO 2discharged to 1 V[20] (Copyright 2016, Springer Nature). (e) Discharge galvanostatic intermittent titration technique (GITT) profiles of the Zn||α-MnO2 cell with the morphology of α-MnO 2 inserted[28] (Copyright 2017, American Chemical Society). (f) Galvanostatic charge/discharge (GCD) profiles within 100 cycles and (g) schematic illustration of electrochemical mechanism of ε-MnO 2 in 1 mol/L ZnSO4+1 mol/L MnSO4 electrolyte[31] (Copyright 2019, Wiley-VCH) |
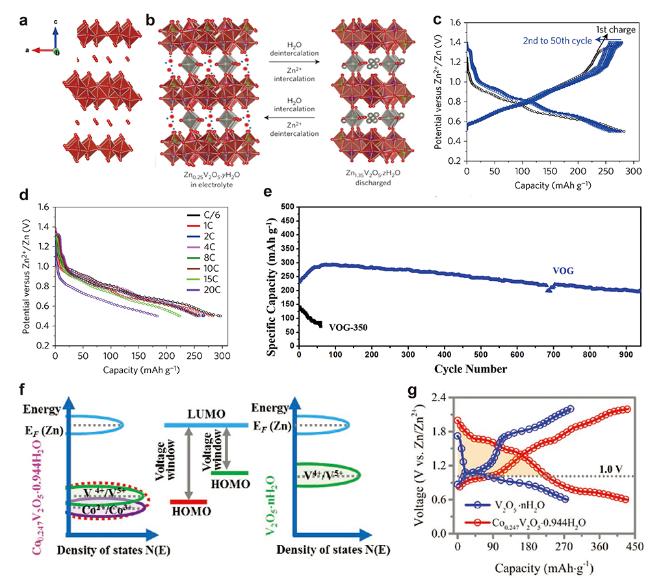
图4 (a) 层状V2O5晶体结构图. Zn0.25V2O5•nH2O正极材料(b)工作机理示意图, (c) 1C (300 mA/g)下GCD图和(d) 4C~20C下倍率放电性能图[38] (Copyright 2016, Springer Nature). (e) VOG(V2O5•nH2O/石墨)和VOG-350(VOG在350 ℃热处理2 h)在6 A/g条件下的循环性能图[39] (Copyright 2018, Wiley-VCH). Co0.247V2O5•0.944H2O和V2O5•nH2O正极材料的(f) 能量态密度示意图和(g) GCD放电曲线对比图[40] (Copyright 2019, Wiley-VCH)Figure 4 (a) Crystal structure of layered V2O5. (b) Schematic illustration mechanism, (c) GCD profiles at a 1C (300 mA/g), and (d) discharge profiles at 4C to 20C rates of Zn0.25V2O5•nH2O cathode[38] (Copyright 2016, Springer Nature). (e) Cycling performance of VOG (V2O5•nH2O/graphene) and VOG-350 (annealing VOG at 350 ℃ for 2 h) at 6 A/g[39] (Copyright 2018, Wiley-VCH). (f) Schematic illustration of the energy versus DOS and (g) GCD profiles comparison of Co0.247V2O5•0.944H2O and V2O5•nH2O cathodes[40] (Copyright 2019, Wiley-VCH) |
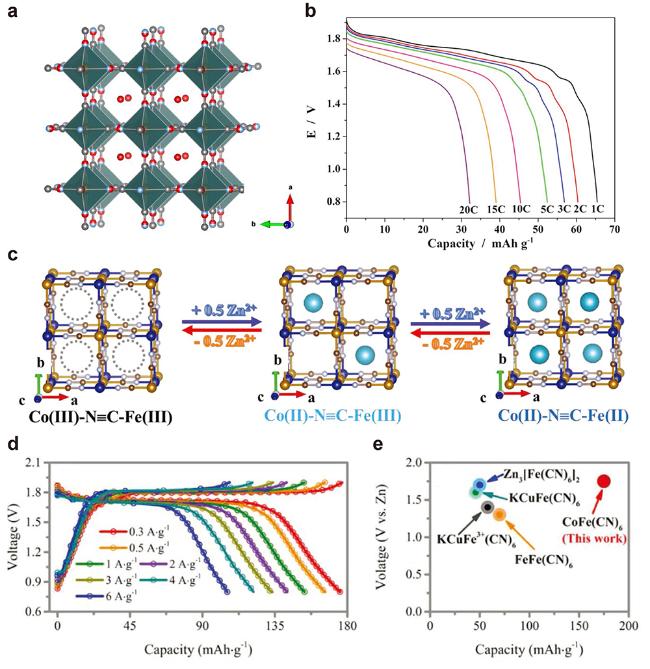
图5 (a) 金属铁氰化物(MHCFs)晶体结构图. (b) Zn3[Fe(CN)6]]2 (ZnHCF)在1 mol/L ZnSO4电解液中的倍率放电曲线[64] (Copyright 2015, Wiley-VCH). (c) 电化学过程中Zn2+可逆嵌入和脱出CoFe(CN)6结构示意图, (d) CoFe(CN)6在不同电流密度下的GCD曲线和(e) CoFe(CN)6与其他MHCFs作为AZIBs正极材料的容量和电压对比 图[66] (Copyright 2019, Wiley-VCH)Figure 5 (a) Crystal structure of the metal hexacyanoferrates (MHCFs). (b) Rate discharge curves of Zn3[Fe(CN)6]]2 (ZnHCF) in 1 mol/L ZnSO4 electrolyte[64] (Copyright 2015, Wiley-VCH). (c) Schematic illustration of reversible Zn2+ intercalation/deintercalation during electrochemical process of CoFe(CN)6, (d) GCD profiles at different current densities of CoFe(CN)6, and (e) comparison of capacity versus voltage with reported MHCFs cathodes for AZIBs of CoFe(CN)6 [66] (Copyright 2019, Wiley-VCH) |
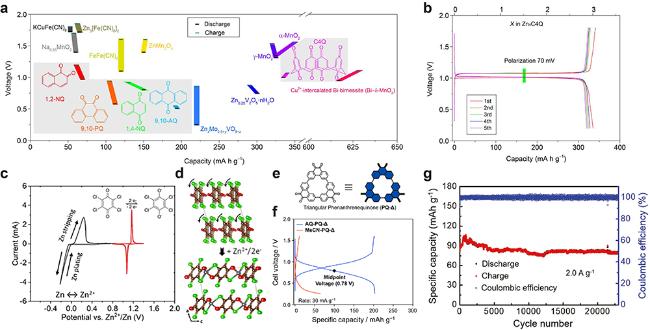
图6 (a) 醌类有机化合物在3 mol/kg Zn(CF3SO3)2电解液中充放电电压和容量与部分报道的正极材料的对比图. (b) C4Q/Zn电池在20 mA/g电流密度下的充放电曲线[74] (Copyright 2018, American Association for the Advancement of Science). (c) 负极Zn和正极四氯苯醌在1 mol/L Zn(CF3SO3)2电解液中的循环伏安曲线(CV). (d) Zn2+嵌入四氯苯醌结构变化模型图[75] (Copyright 2018, American Chemical Society). (e) PQ-△结构式, (f) PQ-△在0.25 mol/L Zn(CF3SO3)2有机电解液(MeCN乙腈溶剂)和3 mol/L Zn(CF3SO3)2水系电解液中GCD充放电曲线对比图[76] (Copyright 2020, American Chemical Society). (g) Zn||DTT电池长循环性能图[78] (Copyright 2018, Wiley-VCH)Figure 6 (a) Discharge/charge voltages and capacities of selected quinone compounds in 3 mol/kg Zn(CF3SO3)2 electrolyte, compared with some reported cathodes. (b) Galvanostatic discharge/charge curves of C4Q/Zn battery at the current density of 20 mA/g[74] (Copyright 2018, American Association for the Advancement of Science). (c) Cyclic voltammograms (CV) of the Zn anode against the p-chloranil cathode in 1 mol/L Zn(CF3SO3)2 electrolyte. (d) Structural change models of p-chloranil upon Zn2+ insertion[75] (Copyright 2018, American Chemical Society). (e) Structural formula and (f) comparison of GCD files of PQ-△ using 0.25 mol/L Zn(CF3SO3)2 organic (MeCN solvent) and 3 mol/L Zn(CF3SO3)2 aqueous electrolytes of PQ-△[76] (Copyright 2020, American Chemical Society). (g) Long-term cycling performance of Zn||DTT battery[78] (Copyright 2018, Wiley-VCH) |
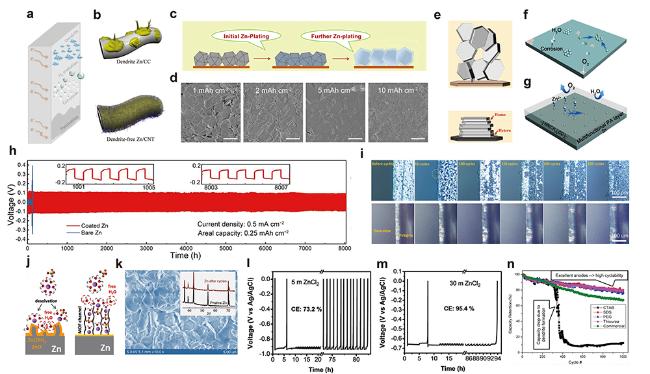
图7 (a) 水系电解液中Zn负极存在问题示意图. (b) Zn在CC和CNTs电极表面沉积示意图[88] (Copyright 2019, Wiley-VCH). (c) Zn在ZIF-8沉积示意图, (d) 在1.0 mA/cm2电流密度下ZIF-8沉积不同容量时Zn的SEM图[89] (Copyright 2019, Elsevier). (e) 外延金属Zn电沉积的设计原理示意图[92] (Copyright 2019, American Association for the Advancement of Science). 空白(f)和PA涂覆(g)后负极Zn沉积示意图, (h) Zn对称电池长周期恒电流循环寿命图[96] (Copyright 2019, Elsevier). (i) Zn和PVB@Zn电极前表面原位光学显微镜图像[99] (Copyright 2020, Wiley-VCH). (j) Zn2+-H2O离子在空白Zn和MOF涂层表面去溶剂化过程示意图[103] (Copyright 2020, Wiley-VCH). (k) Zn负极在1 mol/kg Zn(TFSI)2+20 mol/kg LiTFSI电解液中析出/沉积500周后SEM和XRD图[104] (Copyright 2018, Springer Nature). 在(l) 5 mol/kg和(m) 30 mol/kg ZnCl2电解液中Zn/Zn对称电池中Zn沉积/剥离库伦效率[105] (Copyright 2018, Royal Society of Chemistry). (n) 电解液中有机添加剂对Zn负极循环性能影响[108] (Copyright 2017, American Chemical Society)Figure 7 (a) Schematic diagram of the issues for Zn anodes in aqueous electrolytes. (b) Schematic illustrations of Zn deposition on CC and CNT electrodes[88] (Copyright 2019, Wiley-VCH). (c) Schematic illustration of the Zn plating on ZIF-8, and (d) SEM images of Zn deposits at a current density of 1.0 mA/cm2 for different capacities on ZIF-8[89] (Copyright 2019, Elsevier). (e) Scheme illustrating the design principle of epitaxial zinc metal electrodeposition[92] (Copyright 2019, American Association for the Advancement of Science). Schematic diagrams for Zn deposition on a bare (f) and the PA layer coating (g) electrodes, and (h) long-term galvanostatic cycling of symmetrical Zn cells[96](Copyright 2019, Elsevier). (i) In situ optical microscope images of the front surfaces of Zn and PVB@Zn electrodes[99] (Copyright 2020, Wiley-VCH). (j) Schematic illustration of desolvation process of Zn2+-H2O ion on bare Zn and coating anodes[103] (Copyright 2020, Wiley-VCH). (k) SEM image and XRD pattern (inset) of a Zn anode after 500 stripping/plating cycles in 1 mol/kg Zn(TFSI)2+20 mol/kg LiTFSI electrolyte[104] (Copyright 2018, Springer Nature). CEs of Zn plating/stripping in asymmetric Zn/Zn cells in (l) 5 mol/kg and (m) 30 mol/kg ZnCl2 electrolytes[105] (Copyright 2018, Royal Society of Chemistry). (n) Impact of organic additives in electrolyte on cyclability of zinc anode[108] (Copyright 2017, American Chemical Society) |
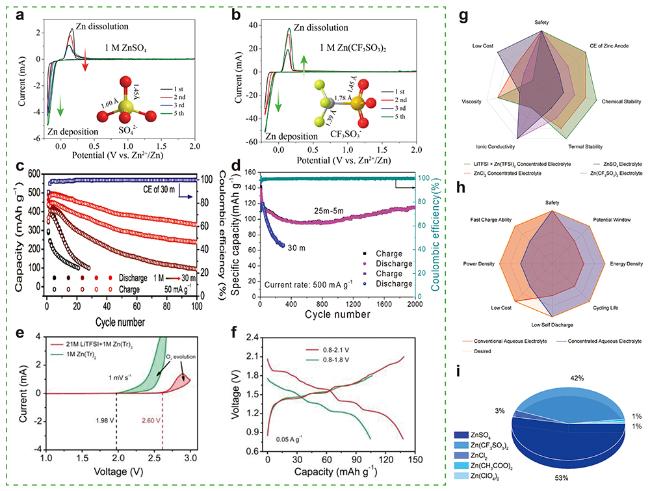
图8 Zn电极在(a) 1 mol/L ZnSO4和(b) 1 mol/L Zn(CF3SO3)2水系电解液中CV图[114] (Copyright 2016, American Chemical Society). (c) Ca0.20V2O5•0.80H2O正极材料在不同浓度ZnCl2电解液中的循环稳定性[45] (Copyright 2019, Wiley-VCH). (d) Na3V2(PO4)2O2F电极材料在25 mol/kg ZnCl2+5 mol/kg NH4Cl和30 mol/kg ZnCl2电解液中的循环稳定性[121](Copyright 2020, Wiley-VCH). 不同电解液中(e) Zn电极的CV图和(f) Zn/VOPO4电池第2周充放电曲线图[72] (Copyright 2019, Wiley-VCH). (g)目前常用传统电解液和高浓度电解液雷达对比图. (h) 传统电解液和高浓度电解液与理想水系电解液性能雷达对比图. (i) AZIBs电解液使用情况Figure 8 CV of Zn electrode in (a) 1 mol/L ZnSO4 and (b) 1 mol/L Zn(CF3SO3)2 aqueous electrolyte[114] (Copyright 2016, American Chemical Society). (c) Cycling stability of Ca0.20V2O5•0.80H2O cathode in different concentrated ZnCl2 electrolytes[45] (Copyright 2019, Wiley-VCH). (d) The cycling stability of the Na3V2(PO4)2O2F electrode in the 25 mol/kg ZnCl2+5 mol/kg NH4Cl and 30 mol/kg ZnCl2 electrolytes[121] (Copyright 2020, Wiley-VCH). (e) CV curves of Zn electrodes and (f) second GCD curves of Zn/VOPO4 batteries in different electrolytes[72] (Copyright 2019, Wiley-VCH). (g) Radar chart comparison of common used conventional and concentrated aqueous electrolytes. (h) Radar chart comparison of the performance of common used conventional and concentrated aqueous electrolytes with desired aqueous electrolyte. (i) The usage of electrolytes of AZIBs |
| [1] |
Armand M. ; Tarascon J.M. Nature 2008, 451, 652.
|
| [2] |
Ji X.L. Energ. Environ. Sci. 2019, 12, 3203.
|
| [3] |
Dunn B. ; Kamath H. ; Tarascon J.M. Science 2011, 334, 928.
|
| [4] |
Soloveichik G.L. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 503.
|
| [5] |
Liu J. ; Hu J.P. ; Deng Q. ; Mo J. ; Xie H. ; Liu Z.C. ; Xiong Y.F. ; Wu X.W. ; Wu Y.P. Isr. J. Chem. 2015, 55, 521.
|
| [6] |
Etacheri V. ; Marom R. ; Elazari R. ; Salitra G. ; Aurbach D. Energ. Environ. Sci. 2011, 4, 3243.
|
| [7] |
Fang G.Z. ; Zhou J. ; Pan A.Q. ; Liang S.Q. ACS Energy Lett. 2018, 3, 2480.
|
| [8] |
Xu W.W. ; Wang Y. Nano-Micro Lett. 2019, 11, 90.
|
| [9] |
Tang B.Y. ; Shan L.T. ; Liang S.Q. ; Zhou J. Energ. Environ. Sci. 2019, 12, 3288.
|
| [10] |
Mai L.Q. ; Mei Z.W. ; Yan M.Y. ; Chen L.N. J. Inorg. Mater. 2017, 32, 225.
|
| [11] |
Kundu D. ; Hosseini Vajargah S. ; Wan L.W. ; Adams B. ; Prendergast D. ; Nazar L.F. Energ. Environ. Sci. 2018, 11, 881.
|
| [12] |
Shoji T. ; Yamamoto T. J. Electroanal. Chem. 1993, 362, 153.
|
| [13] |
Shoji T. ; Hishinuma M. ; Yamamoto T. J. Appl. Electrochem. 1988, 18, 521.
|
| [14] |
Xu C.J. ; Li B.H. ; Du H.D. ; Kang F.Y. Angew. Chem., Int. Ed. 2012, 51, 933.
|
| [15] |
He P. ; Chen Q. ; Yan M.Y. ; Xu X. ; Zhou L. ; Mai L.Q. ; Nan C.W. EnergyChem 2019, 1, 100022.
|
| [16] |
Konarov A. ; Voronina N. ; Jo J.H. ; Bakenov Z. ; Sun Y.K. ; Myung S.T. ACS Energy Lett. 2018, 3, 2620.
|
| [17] |
Zhao Y.L. ; Zhu Y.H. ; Zhang X.B. InfoMat 2020, 2, 237.
|
| [18] |
Wang D.H. ; Wang L.F. ; Liang G.J. ; Li H.F. ; Liu Z.X. ; Tang Z.J. ; Liang J.B. ; Zhi C.Y. ACS Nano 2019, 13, 10643.
|
| [19] |
Huang J.H. ; Wang Z. ; Hou M.Y. ; Dong X.L. ; Liu Y. ; Wang Y.G. ; Xia Y.Y. Nat. Commun. 2018, 9, 2906.
|
| [20] |
Pan H.L. ; Shao Y.Y. ; Yan P.F. ; Cheng Y.W. ; Han K.S. ; Nie Z.M. ; Wang C.M. ; Yang J.H. ; Li X.L. ; Bhattacharya P. ; Mueller K.T. ; Liu J. Nat. Energy 2016, 1, 16039.
|
| [21] |
Guo X. ; Zhou J. ; Bai C. ; Li X. ; Fang G. ; Liang S. Mater.Today Energy 2020, 16, 100396.
|
| [22] |
Xiong T. ; Yu Z.G. ; Wu H.J. ; Du Y.H. ; Xie Q.D. ; Chen J.S. ; Zhang Y.W. ; Pennycook S.J. ; Lee W.S.V. ; Xue J.M. Adv. Energy Mater. 2019, 9, 1803815.
|
| [23] |
Lee B. ; Lee H.R. ; Kim H. ; Chung K.Y. ; Cho B.W. ; Oh S.H. Chem. Commun. 2015, 51, 9265.
|
| [24] |
Zhang N. ; Cheng F.Y. ; Liu J.X. ; Wang L.B. ; Long X.H. ; Liu X.S. ; Li F.J. ; Chen J. Nat. Commun. 2017, 8, 405.
|
| [25] |
Guo C. ; Liu H.M. ; Li J.F. ; Hou Z.G. ; Liang J.W. ; Zhou J. ; Zhu Y.C. ; Qian Y.T. Electrochim. Acta 2019, 304, 370.
|
| [26] |
Jiang Y.Q. ; Ba D.L. ; Li Y.Y. ; Liu J.P. Adv. Sci. 2020, 7, 1902795.
|
| [27] |
Alfaruqi M.H. ; Mathew V. ; Gim J. ; Kim S.J. ; Song J.J. ; Baboo J.P. ; Choi S.H. ; Kim J. Chem. Mater. 2015, 27, 3609.
|
| [28] |
Sun W. ; Wang F. ; Hou S. ; Yang C.Y. ; Fan X.L. ; Ma Z.H. ; Gao T. ; Han F.D. ; Hu R.Z. ; Zhu M. ; Wang C.S. J. Am. Chem. Soc. 2017, 139, 9775.
|
| [29] |
Huang Y.F. ; Mou J. ; Liu W.H. ; Wang X.L. ; Dong L.B. ; Kang F.Y. ; Xu C.J. Nano-Micro Lett. 2019, 11, 49.
|
| [30] |
Jin Y. ; Zou L.F. ; Liu L.L. ; Engelhard M.H. ; Patel R.L. ; Nie Z.M. ; Han K.S. ; Shao Y.Y. ; Wang C.M. ; Zhu J. ; Pan H.L. ; Liu J. Adv. Mater. 2019, 31, e1900567.
|
| [31] |
Chao D.L. ; Zhou W.H. ; Ye C. ; Zhang Q.H. ; Chen Y.G. ; Gu L. ; Davey K. ; Qiao S.Z. Angew. Chem., Int. Ed. 2019, 58, 7823.
|
| [32] |
Guo X. ; Zhou J. ; Bai C.L. ; Li X.K. ; Fang G.Z. ; Liang S.Q. Mater. Today Energy 2020, 16, 100396.
|
| [33] |
Liang G.J. ; Mo F.N. ; Li H.F. ; Tang Z.J. ; Liu Z.X. ; Wang D.H. ; Yang Q. ; Ma L.T. ; Zhi C.Y. Adv. Energy Mater. 2019, 9, 1901838.
|
| [34] |
Zhong C. ; Liu B. ; Ding J. ; Liu X.R. ; Zhong Y.W. ; Li Y. ; Sun C.B. ; Han X.P. ; Deng Y.D. ; Zhao N.Q. ; Hu W.B. Nat. Energy 2020, 5, 440.
|
| [35] |
Kundu D. ; Adams B.D. ; Duffort V. ; Vajargah S.H. ; Nazar L.F. Nat. Energy 2016, 1, 16119.
|
| [36] |
Yang Y.Q. ; Tang Y. ; Fang G.Z. ; Shan L.T. ; Guo J.S. ; Zhang W.Y. ; Wang C. ; Wang L.B. ; Zhou J. ; Liang S.Q. Energ. Environ. Sci. 2018, 11, 3157.
|
| [37] |
Ming F.W. ; Liang H.F. ; Lei Y.J. ; Kandambeth S. ; Eddaoudi M. ; Alshareef H.N. ACS Energy Lett. 2018, 3, 2602.
|
| [38] |
Kundu D. ; Adams B.D. ; Duffort V. ; Vajargah S.H. ; Nazar L.F. Nature Energy 2016, 1, 16119.
|
| [39] |
Yan M.Y. ; He P. ; Chen Y. ; Wang S.Y. ; Wei Q.L. ; Zhao K.N. ; Xu X. ; An Q.Y. ; Shuang Y. ; Shao Y.Y. ; Mueller K.T. ; Mai L.Q. ; Liu J. ; Yang J.H. Adv. Mater. 2018, 30, 1703725.
|
| [40] |
Ma L.T. ; Li N. ; Long C.B. ; Dong B.B. ; Fang D.L. ; Liu Z.X. ; Zhao Y.W. ; Li X.L. ; Fan J. ; Chen S.M. Adv. Funct. Mater. 2019, 29, 1906142.
|
| [41] |
Xia C. ; Guo J. ; Li P. ; Zhang X.X. ; Alshareef H.N. Angew. Chem., Int. Ed. 2018, 57, 3943.
|
| [42] |
Bin D. ; Liu Y. ; Yang B.B. ; Huang J.H. ; Dong X.L. ; Zhang X. ; Wang Y.G. ; Xia Y.Y. ACS Appl. Mater. Inter. 2019, 11, 20796.
|
| [43] |
Du M. ; Liu C.F. ; Zhang F. ; Dong W.T. ; Zhang X.F. ; Sang Y.H. ; Wang J.J. ; Guo Y.G. ; Liu H. ; Wang S.H. Adv. Sci. 2020, 7, 2000083.
|
| [44] |
Liu S. ; Zhu H. ; Zhang B. ; Li G. ; Zhu H. ; Ren Y. ; Geng H. ; Yang Y. ; Liu Q. ; Li C.C. Adv. Mater. 2020, 32, 2001113.
|
| [45] |
Zhang L. ; Rodríguez-Pérez I.A. ; Jiang H. ; Zhang C. ; Leonard D.P. ; Guo Q.B. ; Wang W.F. ; Han S.M. ; Wang L.M. ; Ji X.L. Adv. Funct. Mater. 2019, 25, 1902653.
|
| [46] |
Chen L.N. ; Ruan Y.S. ; Zhang G.B. ; Wei Q.L. ; Jiang Y.L. ; Xiong T.F. ; He P. ; Yang W. ; Yan M.Y. ; An Q.Y. ; Mai L.Q. Chem. Mater. 2019, 31, 699.
|
| [47] |
Liu Y. ; Hu P. ; Liu H. ; Wu X. ; Zhi C. Mater. Today Energy 2020, 17, 100431.
|
| [48] |
Li Z.Q. ; Ren Y.K. ; Mo L. ; Liu C.F. ; Hsu K. ; Ding Y.C. ; Zhang X.X. ; Li X.L. ; Hu L.H. ; Ji D.H. ; Cao G.Z. ACS Nano 2020, 14, 5581.
|
| [49] |
Cao Z.Y. ; Chu H. ; Zhang H. ; Ge Y. ; Clemente R. ; Dong P. ; Wang L.P. ; Shen J.F. ; Ye M.X. ; Ajayan P.M. J. Mater. Chem. A 2019, 7, 25262.
|
| [50] |
Wei T.Y. ; Li Q. ; Yang G.Z. ; Wang C.X. Electrochim. Acta 2018, 287, 60.
|
| [51] |
Alfaruqi M.H. ; Mathew V. ; Song J.J. ; Kim S.J. ; Islam S. ; Pham D.T. ; Jo J. ; Kim S. ; Baboo J.P. ; Xiu Z.L. ; Lee K.S. ; Sun Y.K. ; Kim J. Chem. Mater. 2017, 29, 1684.
|
| [52] |
Hu P. ; Zhu T. ; Wang X. ; Wei X. ; Yan M. ; Li J. ; Luo W. ; Yang W. ; Zhang W. ; Zhou L. ; Zhou Z. ; Mai L. Nano Lett. 2018, 18, 1758.
|
| [53] |
Qian l. ; Wei T.Y. ; Ma K.X. ; Yang G.Z. ; Wang C.X. ACS Appl. Mater. Inter. 2019, 11, 20888.
|
| [54] |
Jiang H.M. ; Zhang Y.F. ; Pan Z.H. ; Xu L. ; Zheng J.Q. ; Gao Z.M. ; Hu T. ; Meng C.G. ; Wang J. Mater. Chem. Front. 2020, 4, 1434.
|
| [55] |
Xia C. ; Guo J. ; Lei Y.J. ; Liang H.F. ; Zhao C. ; Alshareef H.N. Adv. Mater. 2018, 30, 1705580.
|
| [56] |
Chao D.L. ; Zhu C.R. ; Song M. ; Liang P. ; Zhang X. ; Tiep N.H. ; Zhao H.F. ; Wang J. ; Wang R.M. ; Zhang H. ; Fan H.J. Adv. Mater. 2018, 30, 1803181.
|
| [57] |
Wang L.L. ; Huang K.W. ; Chen J.T. ; Zheng J.R. Sci. Adv. 2019, 5, eaax4279.
|
| [58] |
Ding J.W. ; Du Z.G. ; Li B. ; Wang L.Z. ; Wang S.W. ; Gong Y.J. ; Yang S.B. Adv. Mater. 2019, 31, 1904369.
|
| [59] |
Fang G. ; Liang S. ; Chen Z. ; Cui P. ; Zheng X. ; Pan A. ; Lu B. ; Lu X. ; Zhou J. Adv. Funct. Mater. 2019, 29, 1905267.
|
| [60] |
Shan L.T. ; Yang Y.Q. ; Zhang W.Y. ; Chen H.J. ; Fang G.Z. ; Zhou J. ; Liang S.Q. Energy Storage Mater. 2019, 18, 10.
|
| [61] |
de Tacconi N.R. ; Rajeshwar K. ; Lezna R.O. Chem. Mater. 2003, 15, 3046.
|
| [62] |
Paolella A. ; Faure C. ; Timoshevskii V. ; Marras S. ; Bertoni G. ; Guerfi A. ; Vijh A. ; Armand M. ; Zaghib K. J. Mater. Chem. A 2017, 5, 18919.
|
| [63] |
Wang R.Y. ; Shyam B. ; Stone K.H. ; Weker J.N. ; Pasta M. ; Lee H.-W. ; Toney M.F. ; Cui Y. Adv. Energy Mater. 2015, 5, 1401869.
|
| [64] |
Zhang L.Y. ; Chen L. ; Zhou X.F. ; Liu Z.P. Adv. Energy Mater. 2015, 5, 1400930.
|
| [65] |
Trócoli R. ; LaβMantia F. ChemSusChem 2015, 8, 481.
|
| [66] |
Ma L.T. ; Chen S.M. ; Long C.B. ; Li X.L. ; Zhao Y.W. ; Liu Z.X. ; Huang Z.D. ; Dong B.B. ; Zapien J.A. ; Zhi C.Y. Adv. Energy Mater. 2019, 9, 1902446.
|
| [67] |
Ni Q. ; Bai Y. ; Wu F. ; Wu C. Adv. Sci. 2017, 4, 1600275.
|
| [68] |
Li G.L. ; Yang Z. ; Jiang Y. ; Jin C.H. ; Huang W. ; Ding X. ; Huang Y.H. Nano Energy 2016, 25, 211.
|
| [69] |
Ko J.S. ; Paul P.P. ; Wan G. ; Seitzman N. ; DeBlock R.H. ; Dunn B.S. ; Toney M.F. ; Nelson Weker, J. Chem. Mater. 2020, 32, 3028.
|
| [70] |
Wang F. ; Hu E.Y. ; Sun W. ; Gao T. ; Ji X. ; Fan X.L. ; Han F.D. ; Yang X.Q. ; Xu K. ; Wang C.S. Energ. Environ. Sci. 2018, 11, 3168.
|
| [71] |
Shi H.Y. ; Song Y. ; Qin Z. ; Li C. ; Guo D. ; Liu X.X. ; Sun X. Angew. Chem. Int. Ed. 2019, 58, 16057.
|
| [72] |
Wan F. ; Zhang Y. ; Zhang L.L. ; Liu D.B. ; Wang C.D. ; Song L.S. ; Niu Z.Q. ; Chen J. Angew. Chem. Int. Ed. 2019, 58, 7062.
|
| [73] |
Liang Y.L. ; Yao Y. Joule 2018, 2, 1690.
|
| [74] |
Zhao Q. ; Huang W.W. ; Luo Z.Q. ; Liu L.J. ; Lu Y. ; Li Y.X. ; Li L. ; Hu J.Y. ; Ma H. ; Chen J. Sci. Adv. 2018, 4, eaao1761.
|
| [75] |
Kundu D. ; Oberholzer P. ; Glaros C. ; Bouzid A. ; Tervoort E. ; Pasquarello A. ; Niederberger M. Chem. Mater. 2018, 30, 3874.
|
| [76] |
Nam K.W. ; Kim H.J. ; Beldjoudi Y. ; Kwon T.W. ; Kim D.J. ; Stoddart J.F. J. Am. Chem. Soc. 2020, 142, 2541.
|
| [77] |
Wang Y.Y. ; Guo Z.G. ; Ma Y.Y. ; Dong X.L. ; Huang J.H. ; Xia Y.Y. Angew. Chem. Int. Ed. 2018, 57, 11737.
|
| [78] |
Wang Y.R. ; Wang C.X. ; Ni Z.G. ; Gu Y.M. ; Wang B.L. ; Guo Z.W. ; Wang Z. ; Bin D. ; Ma J. ; Wang Y.G. Adv. Mater. 2020, 32, e2000338.
|
| [79] |
Pan H. ; Yu Y.M. ; Bin Z.G. ; Min S.R. ; Neng C.L. ; You A.Q. ; Qiang M.L. Adv. Energy Mater. 2017, 7, 1601920.
|
| [80] |
Li H.F. ; Yang Q. ; Mo F.N. ; Liang G.J. ; Liu Z.X. ; Tang Z.J. ; Ma L.T. ; Liu J. ; Shi Z.C. ; Zhi C.Y. Energy Storage Mater. 2019, 19, 94.
|
| [81] |
Liang H. ; Cao Z. ; Ming F. ; Zhang W. ; Anjum D.H. ; Cui Y. ; Cavallo L. ; Alshareef H.N. Nano Lett 2019, 19, 3199.
|
| [82] |
Zhang N. ; Chen X. ; Yu M. ; Niu Z. ; Cheng F. ; Chen J. Chem. Soc. Rev. 2020, 49, 4203.
|
| [83] |
Li C.P. ; Xie X.S. ; Liang S.Q. ; Zhou J. Energ. Environ. Sci. 2020, 3, 146.
|
| [84] |
Ji X.L. ; Jiang H. Chem. Res. Chinese U. 2020, 36, 55.
|
| [85] |
Chamoun M. ; Hertzberg B.J. ; Gupta T. ; Davies D. ; Bhadra S. ; Van Tassell B. ; Erdonmez C. ; Steingart D.A. Npg Asia Mater. 2015, 7, e178.
|
| [86] |
Parker J.F. ; Chervin C.N. ; Nelson E.S. ; Rolison D.R. ; Long J.W. Energ. Environ. Sci. 2014, 7, 1117.
|
| [87] |
Wang L.P. ; Li N.W. ; Wang T.S. ; Yin Y.X. ; Guo Y.G. ; Wang C.R. Electrochim. Acta 2017, 244, 172.
|
| [88] |
Guo W.B. ; Cong Z.F. ; Guo Z.H. ; Chang C.Y. ; Liang X.Q. ; Liu Y.D. ; Hu W.G. ; Pu X. Energy Storage Mater. 2020, 30, 104.
|
| [89] |
Wang Z. ; Huang J.H. ; Guo Z.W. ; Dong X.L. ; Liu Y. ; Wang Y.G. ; Xia Y.Y. Joule 2019, 3, 1289.
|
| [90] |
Tian Y. ; An Y.L. ; Wei C.L. ; Xi B.J. ; Xiong S.L. ; Feng J.K. ; Qian Y.T. ACS Nano 2019, 13, 11676.
|
| [91] |
Zhang Q. ; Luan J.Y. ; Fu L. ; Wu S.G. ; Tang Y.G. ; Ji X.B. ; Wang H.Y. Angew. Chem. Int. Ed. 2019, 58, 15841.
|
| [92] |
Zheng J.X. ; Zhao Q. ; Tang T. ; Yin J.F. ; Quilty C. ; Renderos G. ; Liu X.T. ; Deng Y. ; Wang L. ; Bock D. ; Jaye C. ; Zhang D.H. ; Takeuchi E. ; Takeuchi K. ; Marschilok A. ; Archer L. Science 2019, 366, 645.
|
| [93] |
Wu C. ; Tan H.T. ; Huang W.J. ; Li W.X. ; Dinh K.N. ; Yan C.S. ; Wei W.F. ; Chen L.B. ; Yan Q.Y. Adv. Funct. Mater. 2020, 30, 2003187.
|
| [94] |
Li H.F. ; Xu C.J. ; Han C.P. ; Chen Y.Y. ; Wei C.G. ; Li B.H. ; Kang F.Y. J. Electrochem. Soc. 2015, 162, A1439.
|
| [95] |
Xia A.L. ; Pu X.M. ; Tao Y.Y. ; Liu H.M. ; Wang Y.G. Appl. Surf. Sci. 2019, 481, 852.
|
| [96] |
Zhao Z.M. ; Zhao J.W. ; Hu Z.L. ; Li J.D. ; Li J.J. ; Zhang Y.J. ; Wang C. ; Cui G.L. Energ. Environ. Sci. 2019, 12, 1938.
|
| [97] |
Kang L.T. ; Cui M.W. ; Jiang F.Y. ; Gao Y.F. ; Luo H.J. ; Liu J.J. ; Liang W. ; Zhi C.Y. Adv. Energy Mater. 2018, 8, 1801090.
|
| [98] |
Zhao K.N. ; Wang C.X. ; Yu Y.H. ; Yan M.Y. ; Wei Q.L. ; He P. ; Dong Y.F. ; Zhang Z.Y. ; Wang X.D. ; Mai L.Q. Adv. Mater. Inter. 2018, 5, 1800848.
|
| [99] |
Hao J.N. ; Li X.L. ; Zhang S.L. ; Yang F.H. ; Zeng X.H. ; Zhang S. ; Bo G.Y. ; Wang C.S. ; Guo Z.P. Adv. Funct. Mater. 2020, 2001263.
|
| [100] |
Hao J.N. ; Li B. ; Li X.L. ; Zeng X.H. ; Zhang S.L. ; Yang F.H. ; Liu S.L. ; Li D. ; Wu C. ; Guo Z.P. Adv. Mater. 2020, 2003021.
|
| [101] |
Cui Y.H. ; Zhao Q.H. ; Wu X.J. ; Chen X. ; Wang Y.T. ; Qin R.Z. ; Ding S.X. ; Song Y.L. ; Wu J.W. ; Yang K. ; Wang Z.J. ; Mei Z.W. ; Song Z.B. ; Wu H. ; Jiang Z.Y. ; Qian G.Y. ; Yang L.Y. ; Pan F. ; L Y. J . Angew. Chem. Int. Ed. 2020, 59, 16594.
|
| [102] |
Yang Q. ; Guo Y. ; Yan B.X. ; Wang C.D. ; Liu Z.X. ; Huang Z.D. ; Wang Y.K. ; Li Y.R. ; Li H.F. ; Song L.S. ; Fan J. ; Zhi C.Y. Adv. Mater. 2020, 32, e2001755.
|
| [103] |
Yang Q. ; Guo Y. ; Yan B.X. ; Wang C.D. ; Liu Z.X. ; Huang Z.D. ; Wang Y.K. ; Li Y.R. ; Li H.F. ; Song L. ; Fan J. ; Zhi C.Y. Adv. Mater. 2020, 32, 2001755.
|
| [104] |
Wang F. ; Borodin O. ; Gao T. ; Fan X.L. ; Sun W. ; Han F.D. ; Faraone A. ; Dura J.A. ; Xu K. ; Wang C.S. Nat. Mater. 2018, 17, 543.
|
| [105] |
Zhang C. ; Holoubek J. ; Wu X.Y. ; Daniyar A. ; Zhu L.D. ; Chen C. ; Leonard D.P. ; Rodríguez-Pérez I.A. ; Jiang J.X. ; Fang C. ; Ji X.L. Chem. Commun. 2018, 54, 14097.
|
| [106] |
BaniβHashemi A. ; Kasiri G. ; Glenneberg J. ; Langer F. ; Kun R. ; LaβMantia F. ChemElectroChem 2018, 5, 2073.
|
| [107] |
Wan F. ; Zhang L.L. ; Dai X. ; Wang X.Y. ; Niu Z.Q. ; Chen J. Nat. Commun. 2018, 9, 1656.
|
| [108] |
Sun K.E. ; Hoang T.K. ; Doan T.N. ; Yu Y. ; Zhu X. ; Tian Y. ; Chen P. ACS Appl. Mater. Inter. 2017, 9, 9681.
|
| [109] |
Mitha A. ; Yazdi A.Z. ; Ahmed M. ; Chen P. ChemElectroChem 2018, 5, 2409.
|
| [110] |
Cai Z. ; Ou Y.T. ; Wang J.D. ; Xiao R. ; Fu L. ; Yuan Z. ; Zhan R.M. ; Sun Y.M. Energy Storage Mater. 2020, 27, 205.
|
| [111] |
Zeng Y.X. ; Zhang X.Y. ; Qin R.F. ; Liu X.Q. ; Fang P.P. ; Zheng D.Z. ; Tong Y.X. ; Lu X.H. Adv. Mater. 2019, 31, 1903675.
|
| [112] |
Yang H.J. ; Chang Z. ; Qiao Y. ; Deng H. ; Mu X.W. ; He P. ; Zhou H.S. Angew. Chem., Int. Ed. 2020, 59, 9377.
|
| [113] |
Song M. ; Tan H. ; Chao D.L. ; Fan H.J. Adv. Funct. Mater. 2018, 28, 1802564.
|
| [114] |
Zhang N. ; Cheng F.Y. ; Liu Y.C. ; Zhao Q. ; Lei K.X. ; Chen C.C. ; Liu X.S. ; Chen J. J. Am. Chem. Soc. 2016, 138, 12894.
|
| [115] |
Huang S. ; Zhu J.C. ; Tian J.L. ; Niu Z.Q. Chemistry 2019, 25, 14480.
|
| [116] |
Kasiri G. ; Trócoli R. ; Bani Hashemi A. ; La Mantia F . Electrochim. Acta 2016, 222, 74.
|
| [117] |
Li W. ; Wang K.L. ; Cheng S.J. ; Jiang K. Energy Storage Mater. 2018, 15, 14.
|
| [118] |
Zhou J. ; Shan L.T. ; Wu Z.X. ; Guo X. ; Fang G.Z. ; Liang S.Q. Chem. Commun. 2018, 54, 4457.
|
| [119] |
Suo L.M. ; Borodin O. ; Gao T. ; Olguin M. ; Ho J. ; Fan X.L. ; Luo C. ; Wang C.S. ; Xu K. Science 2015, 350, 938.
|
| [120] |
Suo L.M. ; Borodin O. ; Wang Y.S. ; Rong X.H. ; Sun W. ; Fan X.L. ; Xu S.Y. ; Schroeder M.A. ; Cresce A.V. ; Wang F. ; Yang C.Y. ; Hu Y.S. ; Xu K. ; Wang C.S. Adv. Energy Mater. 2017, 7, 1701189.
|
| [121] |
Ni Q. ; Jiang H. ; Sandstrom S. ; Bai Y. ; Ren H.X. ; Wu X.Y. ; Guo Q.B. ; Yu D.X. ; Wu C. ; Ji X.L. Adv. Funct. Mater. 2020, 30, 2003511.
|
| [122] |
Hu P. ; Yan M. ; Zhu T. ; Wang X. ; Wei X. ; Li J. ; Zhou L. ; Li Z. ; Chen L. ; Mai L. ACS Appl. Mater. Inter. 2017, 9, 42717.
|
| [123] |
Zhao J.W. ; Li Y.Q. ; Peng X. ; Dong S.M. ; Ma J. ; Cui G.L. ; Chen L.Q. Electrochem. Commun. 2016, 69, 6.
|
| [124] |
Yu P. ; Zeng Y. ; Zhang H. ; Yu M. ; Tong Y. ; Lu X. Small 2019, 15, 1804760.
|
| [125] |
Song W.J. ; Lee S. ; Song G. ; Park S. ACS Energy Lett. 2019, 4, 177.
|
| [126] |
Zhou J. ; Shan L.T. ; Tang B.Y. ; Liang S.Q. Chin. Sci. Bull. 2020, 65, 1. (in Chinese)
周江, 单路通, 唐博雅, 梁叔全, 科学通报, 2020, 65, 1.
|
| [127] |
Zeng Y. ; Zhang X. ; Meng Y. ; Yu M. ; Yi J. ; Wu Y. ; Lu X. ; Tong Y. Adv. Mater. 2017, 29, 1700274.
|
| [128] |
Zhang S. ; Yu N. ; Zeng S. ; Zhou S. ; Chen M. ; Di J. ; Li Q. J. Mater. Chem. A 2018, 6, 12237.
|
| [129] |
Pu S. ; Liao Y. ; Chen K. ; Fu J. ; Zhang S. ; Ge L. ; Conta G. ; Bouzarif S. ; Cheng T. ; Hu X. ; Liu K. ; Chen J. Nano Lett. 2020, 20, 3791.
|
| [130] |
Han Q. ; Chi X. ; Zhang S. ; Liu Y. ; Zhou B. ; Yang J. ; Liu Y. J. Mater. Chem. A 2018, 6, 23046.
|
| [131] |
Li H. ; Liu Z. ; Liang G. ; Huang Y. ; Huang Y. ; Zhu M. ; Pei Z. ; Xue Q. ; Tang Z. ; Wang Y. ; Li B. ; Zhi C. ACS Nano 2018, 12, 3140.
|
| [132] |
Huang S. ; Wan F. ; Bi S. ; Zhu J. ; Niu Z. ; Chen J. Angew. Chem., Int. Ed. 2019, 58, 4313.
|
| [133] |
Mo F. ; Li H. ; Pei Z. ; Liang G. ; Ma L. ; Yang Q. ; Wang D. ; Huang Y. ; Zhi C. Sci. Bull. 2018, 63, 1077.
|
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}