

1 引言
在锂离子电池中, 电极材料扮演着核心角色, 其性能直接决定了电池的容量、充放电速率以及循环稳定 性[5]. 相比于传统的LIB电极材料, 诸如磷酸铁锂(LiFePO4)[6]、镍钴锰氧化物(NMC)[7]、镍钴铝氧化物(NCA)[8]等正极材料, 又如石墨[9]、钛酸锂基[10]、硅基[11]等负极材料. 钒基电极材料凭借较高的理论比容量、较宽的电化学稳定窗口、较低的环境毒性进入研究人员的视野之中[12-13]. 如, Teo等[14]设计了一种锂锡钒氧化物(LiSnVO4)负极材料, 通过溶胶-凝胶法使锂离子电池获得了优异的电化学性能, 在首次不可逆放电容量达到1270 mAh/g后, 在第二次循环结束时可逆容量为305.4 mAh/g, 并在第53次循环结束时仍保持211 mAh/g的容量. De Juan Corpuz等[15]通过水热反应和煅烧制备了多孔ZnV2O4纳米线, 作为锂离子电池负极材料, 该材料在1 A/g电流密度下经100次循环后比容量达460 mAh/g, 在5 A/g电流密度下经1000次循环后比容量为149 mAh/g, 表现出优异的循环稳定性和高比容量. McNulty等[16]通过钠离子交换法从钒氧化物纳米管(VONTs)制备出NaV2O5晶体, 并将其作为锂离子电池负极材料, 从第100个循环到第1000个循环, NaV2O5晶体能够保持其容量的93%, 并且循环后的比容量分别为174 mAh/g和162 mAh/g. Liu等[17]通过第一性原理计算预测了二维V₂N MXene单层作为锂、钠和镁离子电池负极材料的潜力, 对Li、Na、K和Mg离子的扩散能垒分别为0.025、0.014、0.004和0.058 eV, 理论容量分别为925、463和1850 mAh/g, 平均开路电压分别为0.32、0.24和0.34 V. 因此, 如何快速设计并筛选出具有合适电压平台、高理论比容量的新型钒基电极材料成为当前任务.
近年来, 随着材料科学的进步和计算能力的增强, 基于大数据技术的机器学习和第一性原理计算技术在材料研发领域逐渐兴起[18]. 研究者能够以更高的效率筛选和优化电极材料, 进而加速新材料的发现及其性能优化过程[19]. 在新型材料探索方面, Wang等[20]通过机器学习方法预测固态电解质的剪切模量和体积模量, 选择了轻梯度提升机(LGBM)模型作为主要预测模型, 实现了高预测精度并能用于探索新材料. 在预测氧化还原电位方面, Allam等[21]开发了机器学习模型, 以高效准确地预测各种有机材料的氧化还原电位, 并分析了各种分子描述符如何影响氧化还原电位. Jinnouchi等[22]提出了一种结合第一性原理计算和机器学习的方法来预测半电池反应的氧化还原电位, 并通过机器学习力场进行热力学积分, 实现了在广泛相空间上的高效统计抽样, 并通过从粗略近似方法到精确电子结构计算的逐步精细化, 提高了自由能的精度. 对于工作电压预测, Joshi等[23]利用Materials Project数据库中的密度泛函理论(DFT)计算数据, 通过深度神经网络、支持向量回归和核岭回归等机器学习方法来预测电极材料的工作电压. Moses等[24]利用Materials Project数据库的数据训练模型, 通过10折交叉验证评估模型性能, 提出了一种基于深度神经网络回归的机器学习模型, 用于预测金属离子电池电极材料的平均电压(Vav)和充放电过程中的体积变化(ΔV%).
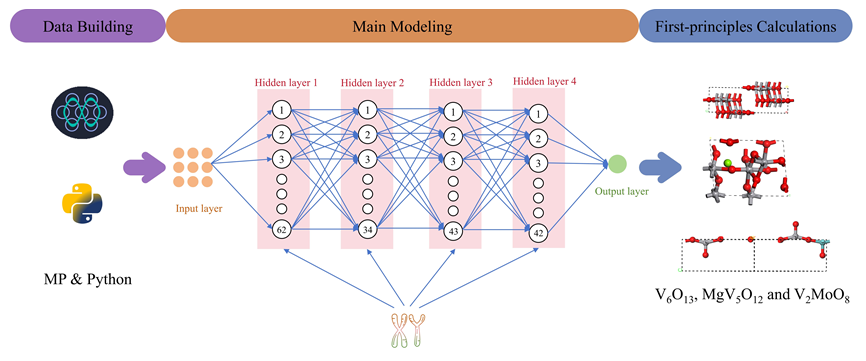
在这项研究中, 构建了一个用于预测钒基电极材料最大理论容量的预测模型, 并通过第一性原理计算进一步验证了预测模型. 为了构建钒基材料数据库, 选取了20种有关材料自身特征作为描述符, 最大理论容量使用pymatgen进行计算, 从而提取出了4694条数据. 为了最大限度减少预测模型预测不准确性, 对数据集进行了噪声处理、归一化处理和相关性处理等特征工程手段. 随后构建了三种机器学习算法用于预测钒基材料, 筛选出26种优异候选钒基电极材料. 经第一性原理计算验证, 确定出V6O13、MgV5O12、V2MoO8三种性能最优的材料.
2 结果与讨论
2.1 数据集分析
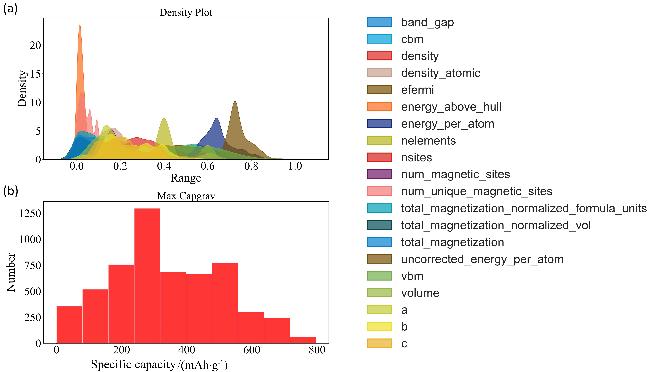
从Materials Project数据库中筛选并提取了共计4694条与钒基材料相关的记录作为本研究的数据集. 通过对前人在锂离子电池电极材料领域的研究成果进行总结, 选定了20种关键特征(解释见支持信息表S1), 这些特征综合覆盖了材料的晶体结构、电子结构及热力学性能等多个维度[27-31]. 从特征分布上可以明显看出, 特征向量的分布差异巨大(支持信息图S1). 为了避免某些特征值过大而导致量级较小的特征被模型低估或忽视的问题, 对数据集中的特征向量实施了归一化处 理[32]. 归一化后的特征向量如图1(a)所示. 值得注意的是, 在归一化处理后, 尽管各个特征向量分布略有不同, 但是大多数服从高斯分布, 这将有利于机器学习模型建立[33]. 但仍有少部分特征不符合高斯分布, 这些非高斯分布的特征向量可能会对模型的学习过程产生影响, 在预测过程中产生异常值, 从而干扰模型的正常工作. 因此, 在后续建模过程中, 需要特别注意选择合适的模型和评估指标, 如深度学习、树类算法、支持向量机模型就适合处理复杂非线性数据, 以确保模型的有效性和鲁棒性[34]. 目标向量分布如图1(b)所示. 大多数钒基材料质量比容量集中在200~300 mAh/g和500~550 mAh/g这两个区间内. 这一现象表明钒基材料相较于工业界常用的磷酸铁锂(LiFePO4: 大约在170 mAh/g左右)以及其他三元材料(指含镍、钴、锰或铝的复合材料: 约200~250 mAh/g)具有显著的优势.
2.2 相关性分析
面对此类多变量优化问题, 本研究利用统计学中的皮尔森相关系数分析法对各特征与目标向量(即材料的比容量)之间的相关性进行了评估[35]. 通过这一分析步骤, 识别并排除了那些与目标向量相关性较低的特征向量, 从而提高模型的解释力和预测精度.
图2为皮尔森相关系数(R值)热力图. 其中对理论容量影响最大的为密度, R值达到0.566, 具有较高线性关系. 在锂离子电池的工作过程中, 锂离子在正极和负极之间的嵌入与脱嵌受到材料本身晶体结构的影响, 而密度对原子堆积方式、结构稳定性、缺陷形成以及相变等产生多方面影响[36]. 密度对锂离子电池的比容量也有着重要影响, 较高的材料密度意味着在相同的体积下可以拥有更多原子数量(图2的nsites相关性与此呼应), 从而提高电池的能量密度, 较高的能量密度通常伴随着较高的比容量[37]. 而材料的最低导带能量、费米能级、最高价带能量与理论容量具有一定相关性, R值分别为–0.238、–0.207、0.235. 最低导带与最高价带的能量水平与材料的氧化还原行为密切相关, 进而影响电池的比容量[38]. 最低导带能量较低的材料更易于接受电子, 促进更有效的氧化还原反应, 从而在锂离子嵌入过程中更有效地储存锂离子. 与之相反, 最高价带能量较高的材料更易于给出电子, 从而在锂离子脱嵌过程中更有效地释放锂离子. 费米能级决定着材料对离子的吸附能力和稳定性, 较低的费米能级意味着更强的电子-离子相互作用, 有利于锂离子的稳定嵌入, 从而提高材料的理论容量[39].
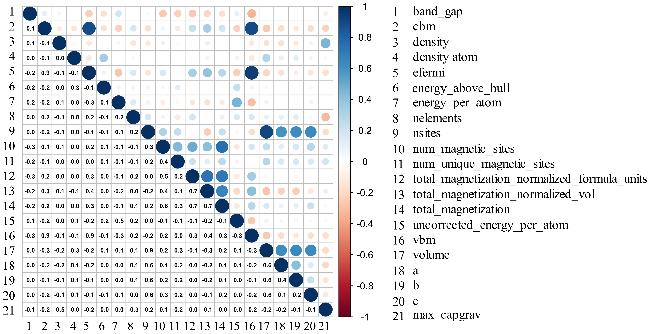
为了进一步探究数据集之间的非线性关系, 将各个特征的皮尔森相关系数的绝对值和互信息的值取平均(R值∈[–1,1], MI值∈[0,+∞]), 得到一个综合特征重要性评分, 如支持信息表S2所示. 相关性分析揭示, 在电极材料的设计中, 材料的密度应该是一个关键的考虑因素. 具体来说, 密度与理论容量之间表现出较强的依赖关系, 这由综合值为0.905所表明. 此外, 在随后的机器学习模型拟合过程中, 应该移除与目标变量相关性较弱的变量(综合评分小于0.1, 表S1中说明被移除特征), 以增强模型的预测稳定性和泛化能力[40]. 而且, 大多数特征向量与目标向量之间的相关性都很弱, 简单的线性机器学习模型不足以完全捕捉这些复杂的关系[41]. 这与数据集分析相谋和.
2.3 模型分析
根据热力图结果显示, 密度对钒基材料比容量正相关性较高. 为了量化评估机器学习模型在预测钒基材料比容量时材料密度及其它输入特征所起到的综合效果, 通过在独立测试集上的决定系数(R2)开展每个算法模型的评估, 并利用最优模型测试集的shap值解释算法模型, 分析各输入特征对模型预测贡献值. 在相关性分析、模型评价、shap解释综合评判下筛选出26种钒基候选电极材料.
图3(a)为各个算法5折交叉验证平均打分. 从图中可以看出, 性能最佳的是深度神经网络算法. 值得说明的是, DNN模型的R2值为0.771, 已经具有实用性与有效性[42]. 数据驱动部分的研究重点在于筛选出对理论容量产生最大影响的关键特征, 通过相关性分析和之后的SHAP分析双层验证, 已经能够明确影响理论容量的关键特征. 通过遗传算法进行超参数优化后, 确定了采用四层隐藏层的设计最为优越, 各层包含的神经元数量分别为62、34、43和42. 此外, 学习率衰减最终被设定为0.00001. 其次, 表现优异的是随机森林算法, 决策树数量为75、最大深度为5、最大叶节点数设置为30. 支持向量回归算法的C(惩罚因子)与epsilon均设置为0.5、核函数选择径向基核(RBF)、gamma参数选择0.02, 并且其R²分数达到了0.659. 图3(b)、(c)和(d)分别为DNN、RF、SVR算法的实际值与预测值之间的对应关系. 横轴代表实际值, 而纵轴则表示预测值. 整体来看, 三种算法预测值与实际值的对应关系主要集中在两个容量区间内: 200~300 mAh/g和500~600 mAh/g. 这与之前对数据集的分析结果相符, 并进一步证实了模型预测的准确性. 深度神经网络(DNN)算法的点分布最为接近回归线y=x, 这表明其预测效果最佳. 但是在600 mAh/g之后预测效果相对于低范围时下降, 这可能是训练集在600 mAh/g之后的样本点稀少的原因, 在之后利用分析结果筛选材料时应注意这个现象. 随机森林(RF)算法大多数数据点都集中在拟合线附近, 但小于200 mAh/g以及大于600 mAh/g的值出现偏差, 与DNN算法类似, 偏差出现原因在于数据分布不均匀以及数据中非线性关系较为复杂, 也说明了钒基材料数据的高维特征之间交互性较强[43]. 支持向量回归(SVR)算法的R2值低于其他两种算法, 表明在这三个模型中, SVR在预测任务上的表现相对较差. 其原因在于, SVR是一种基于边界的回归方法, 试图在保证一定边界内预测误差最小的情况下, 找到一个尽可能平滑的回归超平面[44]. 此外, 鉴于所选取的特征(如密度、电子结构相关参数等)是从材料的晶体结构、电子结构及热力学性能等多维度综合考量而确定的, 同时所采用的三种机器学习算法均为通用的数据处理和模型解释手段, 也能够为不同材料体系的研究提供相似的分析框架, 如钴基、锰基体系.

图3 对钒基材料预测比容量的机器学习算法性能评估. (a)三种算法平均R2; DNN (b)、RF (c)、SVR (d)真实值与预测值对应关系Figure 3 Performance evaluation of machine learning algorithms for predicting specific capacity in vanadium-based materials. (a) Average R2 scores of the three algorithms, (b) actual vs. predicted values for DNN, (c) actual vs. predicted values for RF, and (d) actual vs. predicted values for SVR |
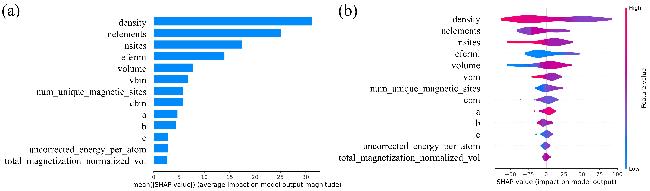
图4(a)与(b)展示了深度神经网络算法中经相关性筛选之后各个特征的SHAP分析结果[45]. 4(a)为特征重要性的分布图, 这是一种直观展示各个特征对模型预测影响程度的工具. 在先前的相关性研究环节, 已经通过统计分析确定了各个特征与理论容量之间的相关性. 为了进一步验证这些发现, 采用了SHAP值(SHapley Additive exPlanations)分析, SHAP值是一种解释模型预测的方法, 基于博弈论中的Shapley值, 能够量化每个特征对模型预测结果的贡献度[46]. 发现材料密度的SHAP平均值显著高于其他特征, 这进一步证明了在数据层面上密度对于理论容量的影响程度较深. 此外, 材料中总元素种类数量(nelements)与材料中原子或分子数量(nsites)这两个特征也具有较高影响. nelements越多, 不同元素的协同作用越复杂. 不同元素的协同作用, 不仅能够提高电导率和化学吸附能力, 还可以保持材料的结构完整性[47]. 较多的nsites, 可以构建出更复杂的结构网络, 从而增加无序结构的可能性, 提供更多的潜在离子迁移路径, 减少了锂离子在传输过程中的阻力[48-49]. 图4(b)展示了各个特征值分布的小提琴图. 从图中可以看出, 密度特征的颜色分布较深, 这表明对于DNN模型的预测具有较高的影响力. 同时, 小提琴图横向形状较宽, 说明在数据集中分布较为广泛, 包含多样化的值, 数据集中的样本具有广泛的变异性[50]. 此外, 在小提琴图中未观察到广泛数据点偏离集中分布区域的现象, 这表明异常值与极端值较少[51].
通过密度这个重要特征进行筛选工作, 选定原始数据集前0.5%的数据, 确定了密度大于3.70的26种钒基材料(支持信息表S3). 如表1所示, 此处展示理论容量最高的三种材料信息, 包括化学式、计算的理论比容量、密度以及在Materials Project数据库中的ID信息, 若要想进一步了解该材料基本信息, 可以根据ID前往官方网站(https://next-gen.materialsproject.org)进行检索. 可以发现, 所筛选的26种候选材料大部分皆为钒氧化物以及钒酸盐材料, 诸如氧化钒、钒酸镁、钒酸钼等. 钒氧化物由于其多价态特性, 在充放电过程中可以发生可逆的氧化还原反应, 从而实现较高的理论容量[52]. 钒酸盐材料则因其层状结构和隧道结构提供了快速的离子扩散通道, 有助于提高材料的倍率性能和循环稳定 性[53]. 这些特性共同作用, 使得钒基材料成为高性能锂离子电池电极材料的理想候选者.
表1 优异性能候选材料基本信息Table 1 Basic information of candidate materials with superior performance |
| ID | Formula | Density/(g•cm–3) | Specific capacity/(mAh•g–1) |
|---|---|---|---|
| mp-18896 | V6O13 | 3.927 | 755.468 |
| mp-2229279 | MgV5O12 | 3.877 | 699.022 |
| mp-27877 | V2MoO8 | 3.755 | 678.126 |
2.4 计算验证
V6O13、MgV5O12和V2MoO8优化之后的晶体结构如图S2. V6O13属单斜C2/m空间群, 含V5+和两种V4+, V—O键长0.166~0.228 nm. MgV5O12属三斜P1空间群, Mg2+与六个O2–形成扭曲八面体, V4+和V3+六配位. MoV2O8亦为单斜C2/m空间群, V5+和Mo6+六配位, Mo—O键长0.189~0.195 nm, 各化合物中O2–配位数及几何形态多样.
图5为三种材料在费米能级附近能带结构、态密度图(图S3为整体图). V6O13 [图5(a)]的能带与费米能级相交, 显示出固有的金属性质[54]. 其态密度图在费米能级附近形成明显峰值, 表明该材料具有较高的电子态密度, 有利于提高导电性, 其中V(d)轨道对电子态密度的贡献尤为显著. MgV5O12 [图5(b)]同样显示了金属特性, 费米能级附近的电子态密度主要由V(s, p, d)轨道贡献, 特别是d轨道, 而O(s, p)和Mg(s, p)轨道也有一定贡献但相对较小. 相比之下, V2MoO8 [图5(c)]的费米能级位于价带顶和导带底之间, 表现出半金属特性[55], O(p)轨道对其电子态密度贡献最大. 总体上, 这三种材料均展现了良好的导电能力, 但V6O13在费米能级附近的电子态密度最高, 显示出更优的导电性能[56]. 图5(d)展示了三种材料的电子密度分布情况, 右侧的颜色条表示相对电子密度, 红色代表较高密度, 蓝色表示较低密度. 对于V6O13, 电子密度主要集中在氧上, 由于锂离子通常是通过与阴离子(在这里主要是氧原子)之间的库仑力进行吸附的, 因此高电子密度区域能够增强锂离子的吸附强度[57]. 此外, 电子云的密集分布可能有助于形成稳定的锂插入位点, 这对于实现高效的充放电循环至关重 要[58]. 针对MgV5O12, 除了氧原子外, 还可以观察到电子密度聚集在镁原子周围, 表明镁的存在会改变局部电子环境, 影响锂离子的吸附位置或方式[59]. 有研究表明, 如果某些电子密度高的区域靠近镁原子, 则可能成为锂离子优先选择的吸附位点[60]. 至于V2MoO8, 虽然电子密度依然主要集中在氧原子上, 但钼原子周围的电子密度不容忽视, 作为过渡金属, 未填满的d轨道能够参与成键并影响电子云的形状, 这可能导致特定的电子密度模式, 使得锂离子更容易找到合适的吸附位点[61].
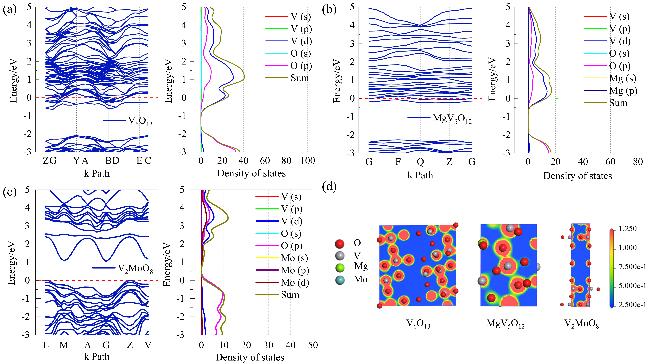
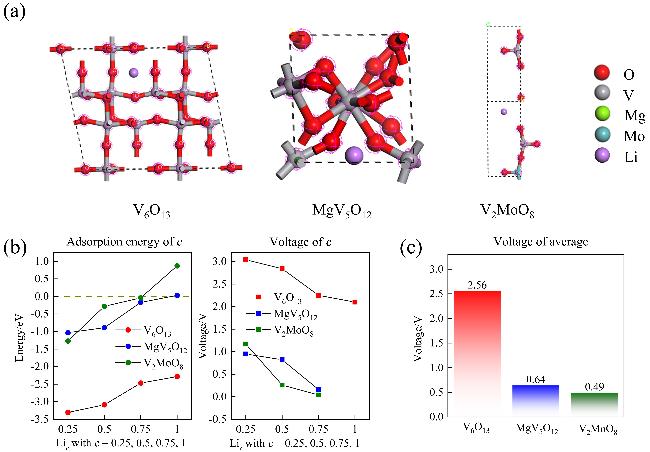
图6为三种材料吸附能及开路电压计算图. 图6(a)为计算出的最低构型, 产生的吸附位点及吸附能如表S4所示. MgV5O12产生的吸附位点为三个, 而V6O13与V2MoO8产生的吸附位点为一个, 可能由于MgV5O12结晶于三斜晶系的P1空间群, 其结构较为复杂, 包含多种不同位置的Mg2+、V4+和V3+, 而V6O13与V2MoO8结晶于单斜晶系的C2/m空间群, 其结构相对简单, 导致了MgV5O12的表面可能存在多种不同的活性位点[62]. 其中V6O13的吸附能相较于MgV5O12与V2MoO8更低, 为–3.304 eV, 可能因为MgV5O12和V2MoO8中的电荷分布相对集中, 特别是在离子键形成的区域, 这种集中的电荷分布使得外来分子难以有效地与表面发生相互作用, 从而降低了吸附能力[63].
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
电极材料的开路电压(OCV)是锂离子电池中的重要参数[17]. 在此构建超胞模型来研究三种电极材料的开路电压, V6O13、MgV5O12和V2MoO8通过构建2×1×2、2×2×1、1×1×4超胞模型来研究开路电压, 通过在之前计算出的三种材料吸附位点上放置不同数目的锂离子, 探索了一系列化学式为MLic (M为V6O13、MgV5O12和V2MoO8)的吸附构型, 其中c=0.25, 0.5, 0.75和1定义为每个化学式MLic中锂离子的数目, c越大, 表明锂离子在MLic上的覆盖率越高(见支持信息图S4、S5、S6). 图6(b)左侧显示了吸附能对c的依赖性. 可以看出, 由于锂离子之间的排斥作用增强, 吸附能随着吸附锂离子数目的增大而减小, V6O13、MgV5O12这种趋势并不明显,吸附能在c从0.25到1分别增长了1.021和1.063 eV, 值得注意的是, MgV5O12在c=1时, 吸附能增长到了0.0296 eV (>0 eV), 表明吸附构型开始变得不稳定[64]. 而V2MoO8增长了2.142 eV, 尤其在c≥0.75时尤为明显, 从–0.042 eV增长到了0.87 eV, 这可能是因为Mo的引入改变了电子的分布, 尤其是Mo的d轨道对总态密度的贡献, 影响了吸附过程中电子的转移和共享, 从而影响吸附能[61]. 图6(b)右侧显示了开路电压随c变化图. 吸附锂离子最大数目对于V6O13Lic是c=1, 而对于MgV5O12Lic和V2MoO8Lic是c=0.75. 因此可以算出锂离子的V6O13、MgV5O12和V2MoO8的平均OCV分别为2.56 V (这与实验值极其相近)[65]、0.64 V、0.49 V, 如图6(c)所示. 经平均OCV计算得出, V6O13适合作为锂离子电池正极材料, 而MgV5O12和V2MoO8适合作为锂离子电池负极材料[17].
3 结论
通过机器学习模型与第一性原理计算构建了一种筛选-验证流程. 从4694条钒基数据中发现了26种潜在的候选材料. 具体而言, 从Materials Project数据库中提取了包含20种关键特征的4694条钒基材料信息, 并使用pymatgen高通量计算以确定每种材料的最大理论比容量. 为了筛选最相关特征向量, 通过皮尔森系数(R)分析了每种特征与比容量的相关性, 并初步筛选出有效特征并排除了冗余特征. 其中, 密度显示出与比容量的高度相关性. 通过对三种不同算法的综合比较, 深度神经网络算法(DNN)获得了最高的预测评分. 利用SHAP解释模型揭示了密度对DNN模型预测结果的贡献度最高. 基于这些发现, 按照密度特征选取原始数据集前0.5%数据, 最终确定了26种有潜力的电极候选材料. 经第一性原理计算验证, 验证了3种理论容量均大于650 mAh/g, 平均电压分别为2.56、0.64、0.49 V的钒基正负两电极候选材料. 这项研究表明, 这一筛选-验证流程对于探索新型钒基候选材料是一种有效的替代方法, 并有可能扩展应用于其他材料的研发领域.
4 实验部分
4.1 算法模型与评估
构建了一种以深度神经网络(DNN)为核心架构的预测模型, 该模型被设计用于与支持向量回归(SVR)、随机森林(RF)算法进行比较, 以确定最优的特征预测模型. 为了进一步优化模型的性能, 应用了遗传算法来调整三种算法超参数(包括但不限于学习率和各隐藏层中神经元的数量). 遗传算法的种群大小与进化代数皆设置为50, 交叉与异变概率分别为0.5与0.2(优化流程参见支持信息S2.4). 为了确保模型评估结果的准确性及增强模型的泛化能力, 采取了5折交叉验证以减少模型对于特定数据集的依赖性[66]. 为了量化模型性能, 选取了决定系数(R2)作为评价标准, 该系数能够直观地反映模型解释变量间关系的能力, 其计算方法如公式(1)所示[67]:
${{R}^{\text{2}}}=\text{1}-\frac{\sum\limits_{i=\text{1}}^{n}{\left( {{y}_{i}}-\hat{y} \right)}}{\sum\limits_{i=\text{1}}^{n}{\left( {{y}_{i}}-\overline{y} \right)}}$
其中yi为目标变量真实值, $\hat{y}$为目标变量预测值, $\bar{y}$为目标变量预测值的平均值.
4.2 第一性原理计算方法
构建了V6O13、MgV5O12、V2MoO8超胞模型, 交换关联函数采用广义梯度近似(GGA)中的Perdew-Burke- Ernzerhof (PBE)泛函处理, 离子核与价电子之间的相互作用通过超软赝势方法处理. V6O13、MgV5O12、V2MoO8的截断能设置皆为571.40 eV; 积分波函数积分点数量(k-point)分别为2×7×3, 5×5×3, 1×7×6. 几何优化过程中采用了Broyden-Fletcher-Goldfarb-Shanno (BFGS)方法. 结构优化过程中, V6O13、MgV5O12、V2MoO8的所有原子位置和晶格参数都进行了完全弛豫.
计算了吸附能, 锂离子在三种材料上的吸附能定义为[68]:
${{E}_{\text{ads}}}=\text{(}{{E}_{\text{total}}}-{{E}_{\text{substrate}}}-c{{E}_{\text{Li}}}\text{)/}c$
其中Etotal是体系吸附之后的总能量, Esubstrate是基底能量, ELi是锂单质的化学势, 并被视为在其体相中每一个客体原子的能量, c是吸附原子数.
此外, 还计算了开路电压与平均开路电压, 这是表征可充电电池输出电压的关键参数, 开路电压公式定义为[17]:
$\text{OCV}=\text{(}{{E}_{\text{M}}}+c{{E}_{\text{Li}}}-{{E}_{\text{ML}{{\text{i}}_{c}}}}\text{)/e}$
这里M表示V6O13、MgV5O12和MoV2O8三种材料, EM, EMLic表示吸附锂离子前后的能量, ELi表示锂单质能量.
(Cheng, F.)