

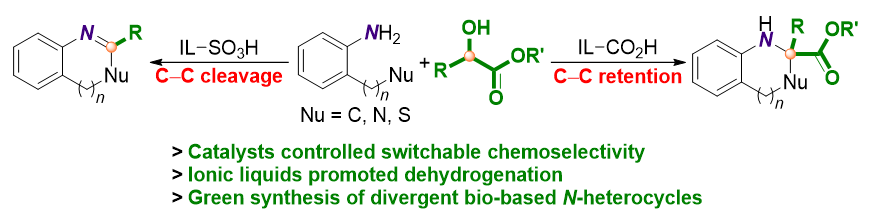
1 引言

α-羟基酯, 如乳酸酯、扁桃酸酯、乙醇酸酯及苹果酸酯等, 是一类新兴的生物质基平台分子, 可以通过纤维素、淀粉以及其他糖类化合物制备[6]. α-羟基酯中的羟基与酸性基团可以参与氧化[7]、消除[8]、胺化[9]以及 C—C键断裂[10]等多种类型的转化, 进而生成高附加值的直链化合物(如酮酯、α,β-不饱和酯、α-卤代酯、酸以及酮等), 然而, 利用此类底物构建杂环化合物的研究却鲜见报道. Wan和Yang团队[11]以乳酸乙酯作为C2或C3合成子, 在金属催化作用下实现了氮杂环的高效合成(Scheme 1b); 我们课题组[12]最近也初步探讨了其作为C1合成子的应用潜力. 鉴于此, 开发基于α-羟基酯的氮杂环的绿色、高效制备方法具有重要的理论和现实意义.
作为α-羟基酯的氧化物, α-酮酸酯可以作为酰基替代物, 在过渡金属、光氧化还原或氧化物催化剂的作用下释放无毒的CO2气体[13], 并作为典型碳合成子通过胺化反应制备多样化的氮杂环[14]. 得益于α-羟基酯良好的可再生特性及优异的溶解性能, 我们推测直接将其作为α-酮酸酯的替代物或许更具前景——既可充当绿色碳源, 又可作为反应介质. 同时, 在脱羧反应中, 酯类较羧酸更为稳定, 因此α-羟基酯可以作为可调碳源参与杂环的多样性制备, 然而, 醇羟基较低的反应活性使得直接胺化反应具有一定的挑战. 研究表明, 在金属-碱体系中, 醇可以通过脱氢或借氢策略暂时转化为反应性更高的醛基[15]. 如果能将其原位转化为高活性的α-酮酸酯, 则将使后续胺化反应的难度大大降低. 因此, 开发一种适用于α-羟基酯的脱氢和选择性脱羧的高效催化体系是实现目标转化的关键所在.
离子液体(ILs)凭借其对极性生物质的良好溶解性以及通过氢键效应对生物质基化合物出色的识别能力, 在生物质转化中应用广泛[16]. 此外, 受离子液体独特的可调性、可回收性及其促进脱羧反应的能力的启发[17], 我们推测通过设计酸性功能化的离子液体催化剂可以精准调控α-羟基酯的C—C键断裂. 氮杂芳环作为药物与农用化学品的关键结构单元, 其重要性可见一斑——在美国食品药品监督管理局(FDA)批准的1086种小分子药物中, 有多达640种属于杂环范畴[18]. 基于Nan和Liu团队[19]对氮杂环合成的持续探索, 本研究以生物质源α-羟基酯为可调的C1合成子, 通过磺酸或羧酸功能化ILs调控C—C键断裂, 实现多种生物质基氮杂环的多样性合成. 具体而言, 羧酸型ILs可以通过脱氢环化构建带有季碳中心的二氢喹喔啉, 其中脱氢是在离子液体的阴离子和阳离子的协同作用下实现的, 与传统的金属-碱催化机制截然不同; 磺酸型ILs则可以通过氢键效应与强固体酸等优势整合, 促进串联脱氢环化与脱羧过程, 实现喹唑啉酮、苯并咪唑、苯并噻唑及喹喔啉等氮杂环的催化制备(Scheme 1c).
2 结果与讨论
2.1 催化体系的优化
我们对乳酸乙酯(2a)与2-吲哚苯胺(1a)的串联催化转化条件进行了探索(Table 1). 鉴于乳酸乙酯的优异溶解性, 反应最初在无外加溶剂的条件下进行. 幸运的是, 在乙酸存在的条件下我们同时检测到了喹喔啉(4aa)与二氢喹喔啉(3aa)的生成(Entry 1); 然而在两种不同的催化体系下, 底物的选择性均不够理想(Entry 2). 考虑到离子液体的化学性质可以通过阳离子、阴离子和取代基的理性设计来精准调控, 我们对酸性功能化的离子液体进行了系统筛选. 值得注意的是, 通过离子液体中酸性官能团的调控, 我们实现了乳酸乙酯的选择性转化. 具体而言, 含弱酸性羟基或羧基的离子液体倾向于生成二氢喹喔啉, 且前者的催化性能明显弱于后者(Entries 4 vs. 3). 磺酸功能化离子液体则可以通过乳酸乙酯的C—C键断裂, 主要生成喹喔啉产物(Entry 5). 基于此, 我们进一步设计了含双酸性位点的离子液体. 令人高兴的是, 其催化性能(包括收率与选择性)较单酸型显著提升(Entries 5~13). 研究表明, 以咪唑为骨架的双功能离子液体表现最佳(Entries 8 vs. 5和6); 连接离子液体的碳链的延长并未使其活性进一步提高(Entries 9 vs. 7); 含双Lewis/Brønsted酸性的离子液体(如Cat-8)可以获得单一选择性的喹喔啉(4aa)产物(Entries 11和12), 其中, 以In(OTf)₄⁻为阴离子的催化剂Cat-8在6 h内可以得到高达90%的4aa分离产率(Entry 12). 值得注意的是, 当催化剂用量降低时仍然可以得到较好的反应结果(Entry 13), 且OTf⁻阴离子的效果略优于OAc⁻ (Entries 3 vs. 15), 而其他的Brønsted酸的催化效果均不甚理想(Entries 17和18). 因此, Cat-8和[HO2CMemim]OTf分别是制备两种产物的最佳催化剂选择(Entries 13和14).
表1 模板底物的反应条件优化aTable 1 Optimization of reaction conditions of model substratesa |
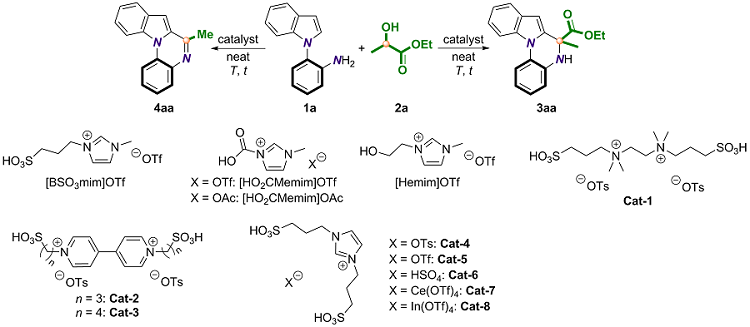
| Entry | Acid (equiv.) | Yield 3aa/% | Yield 4aa/% |
|---|---|---|---|
| 1 | AcOH (1) | 18 | 14 |
| 2 | [BSO3mim]OTf (0.5) | 20 | 40 |
| 3 | [HO2CMemim]OTf (0.5) | 77 | — |
| 4 | [Hemim]OTf (0.5) | 68 | — |
| 5 | Cat-1 (0.5) | — | 56 |
| 6 | Cat-2 (0.5) | 11 | 64 |
| 7 | Cat-3 (0.5) | 7 | 68 |
| 8 | Cat-4 (0.5) | 7 | 76 |
| 9 | Cat-5 (0.5) | 13 | 70 |
| 10 | Cat-6 (0.5) | 9 | 74 |
| 11 | Cat-7 (0.5) | — | 88 |
| 12 | Cat-8 (0.5) | — | 90b |
| 13 | Cat-8 (0.3) | — | 88b (74b,c) |
| 14 | [HO2CMemim]OTf (0.5) | 75 (64c) | — |
| 15 | [HO2CMemim]OAc (0.5) | 71 | — |
| 16 | [HO2CMemim]OTf (0.5) | 72d | 11 |
| 17 | PivOH (1) | 20 | 25 |
| 18 | TsOH (1) | — | 40 |
a Reaction conditions: 1a (0.10 mmol), 2a (0.3 mL), and acid were stirred at 140 ℃ for 12 h under air. b The reaction was conducted for 6 h. c The reaction was carried out at 120 ℃. d The reaction was conducted for 24 h. PivOH: trimethylacetic acid; TsOH: p-toluenesulfonic acid. |
2.2 反应底物的拓展
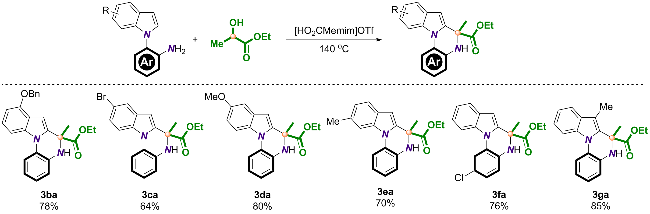
为了验证合成策略的普适性, 我们在最优催化条件下探究了多种2-吲哚苯胺与乳酸乙酯(2a)反应性能(Schemes 2和3). 结果表明, 通过同样的合成路线, α-羟基酯2a可以实现喹喔啉类与二氢喹喔啉类产物的绿色和高效制备. 在[HO2CMemim]OTf的调控作用下, 通过取代基的精准修饰可以获得一系列含季碳中心的二氢喹喔啉(3ba~3ga), 且收率良好. 该反应的电子效应较为显著, 含吸电子基的底物(1c、1f)活性均低于供电子基底物(1b、1d). 值得说明的是, 研究发现空间位阻效应较为反常, C3位甲基取代的吲哚(1g)收率更高, 表明吲哚环的电子特性(而非空间位阻)对结果具有决定性作用 ——环体系亲核性的增强促进了吲哚C2位的分子内进攻, 从而得到目标环化产物.
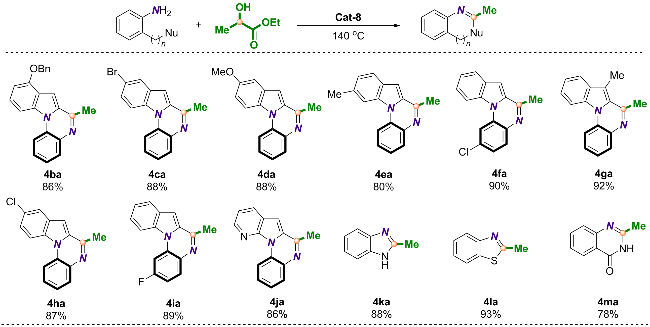
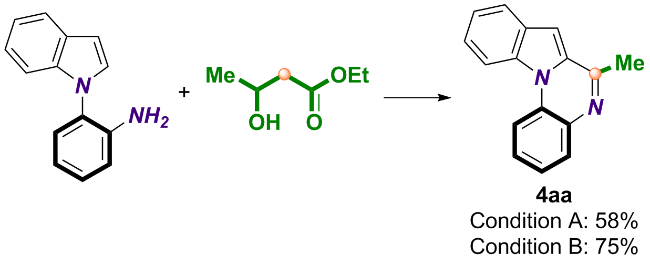
随后, 我们以Cat-8为催化剂探索了喹喔啉类化合物的合成路径(Scheme 3), 通过串联脱羧环化反应以80%~92%的产率高效制备了一系列产物(4ba~4ja). 结果表明, 虽然取代基电子效应对收率影响微弱, 然而富电子的C3位甲基取代底物(1g)仍略有优势. 卤素官能团(如4ca、4fa、4ha、4ia)与7-氮杂吲哚(4ja)均可兼容该体系, 为目标产物的后续衍生化提供了更多可能. 基于吲哚类底物的成功转化, 我们将反应进一步拓展至其他杂环体系, 以苯胺和乳酸乙酯分别作为底物和C1合成子, 实现了喹唑啉酮、苯并咪唑以及苯并噻唑等(4ka~4ma)的高效构筑, 证明了该方法的普适性与多样性.
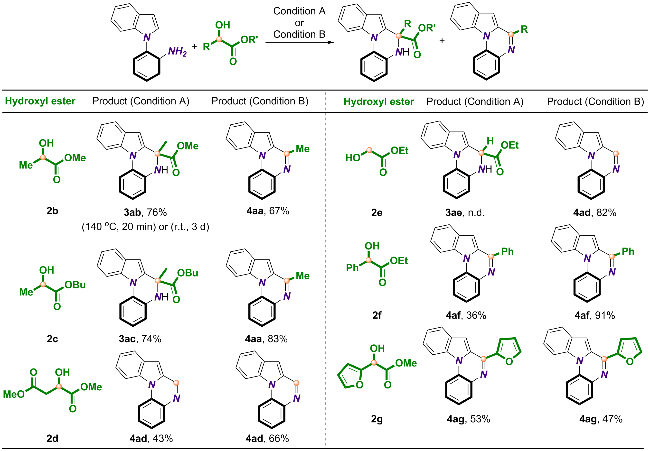
接下来, α-羟基酯的衍生化也证明了该策略的实用性(Scheme 4). 研究表明, 除了乳酸乙酯(2a)外, 乳酸甲酯(2b)与乳酸丁酯(2c)也可以在Cat-8的催化下得到相同的目标产物4aa. 苹果酸二甲酯(2d)则可以通过逆- Aldol反应生成乙醛酸甲酯中间体, 后者进一步与1a发生脱羧环化得到4ad; 相似地, 乙醇酸乙酯(2e)也可以经脱羧环化生成相同的目标产物. 扁桃酸乙酯(2f)与α-羟基-2-呋喃乙酸甲酯(2g)则可以分别将苯基与呋喃基团引入到喹喔啉骨架中(2f→4af, 2g→4ag). 而对于[HO2CMemim]OTf催化体系, 乳酸甲酯(2b)与乳酸丁酯(2c)可以分别以76%和74%的分离产率生成二氢喹喔啉类产物3ab和3ac. 与此相反, 扁桃酸乙酯(2f)和α-羟基-2-呋喃乙酸甲酯(2g)通过C—C键断裂生成喹喔啉4af和4ag, 这可能是由于空间位阻的引入产生了更为拥挤的碳中心原子. 当以苹果酸二甲酯(2d)为底物时, 可以顺利得到4ad, 而对于乙醇酸乙酯(2e), 我们并未检测到任何目标产物的生成.
2.3 模板反应的放大
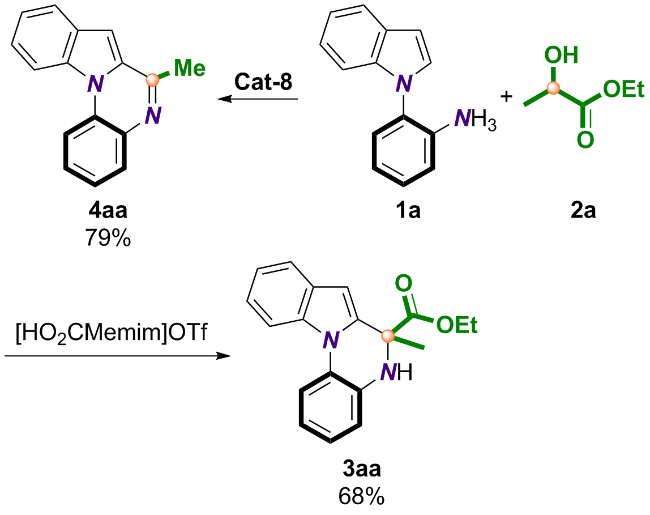
为了进一步论证该生物质基合成策略的实用价值和前景, 我们进行了放大实验(Scheme 5). 结果表明, 在两种催化体系下, 3aa与4aa均能以较高收率实现克级制备.
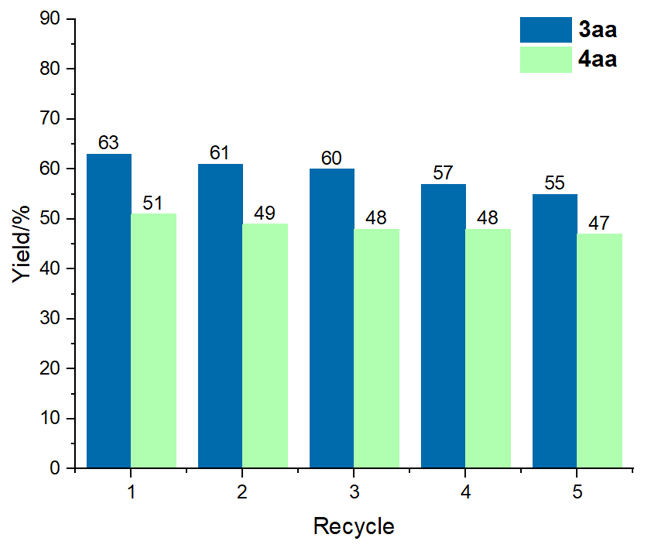
2.4 催化体系的循环性能研究
2.5 反应机理的探究
为阐明差异化合成路径的反应机理, 我们开展了一系列控制实验(Scheme 7). 首先, 在Cat-8催化反应初期, 我们以74%的产率分离得到中间体3aa (Scheme 7a); 3aa可定量转化为4aa (Scheme 7b), 表明3aa可能是喹喔啉合成过程中的关键中间体; N-甲基取代的苯胺(7)无法得到环化产物, 表明伯胺NH2基团对亚胺中间体的形成至关重要(Scheme 7c); 直接使用丙酮酸乙酯(5)作为底物, 可以顺利得到3aa (76%)和4aa (72%)两种目标产物(Scheme 7d); 此外, 当将2a直接与催化剂相互作用时, 可以检测到氢气的释放, 表明该反应可能经历脱氢步骤生成丙酮酸乙酯(5) (Scheme 7e).
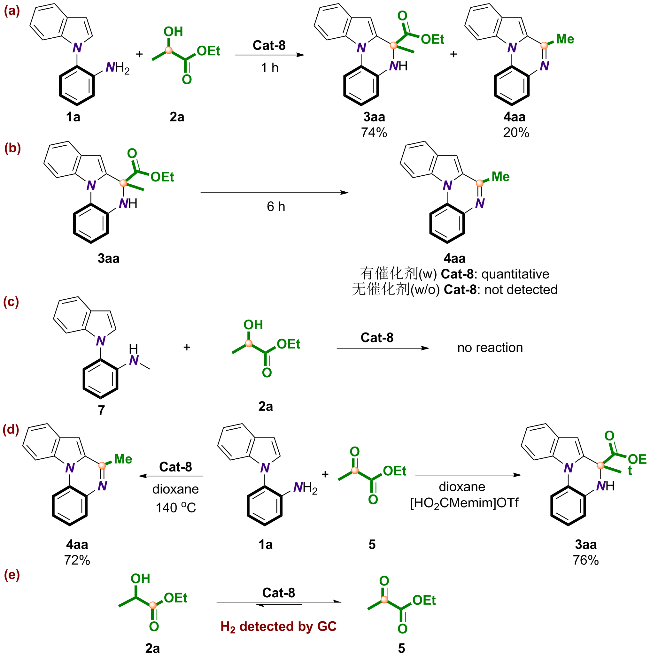
我们进一步研究了催化剂的作用机制与影响因素(Scheme 8). 结果表明, 在不使用离子液体催化剂时, 仅得到微量的产物3aa, 而在无酸性功能基的离子液体作用下, 收率竟高达61%. 在反应初期, 没有催化剂的体系没有检测到任何产物分子, 而[Bmim]BF4与羧酸型离子液体经3 h反应分别可以得到23%和41%的目标产物(Scheme 8a). 在无催化剂情况下, 3aa无法转化为4aa, 而酸性催化剂下可定量生成(Scheme 7b), 表明离子液体中阴阳离子协同作用可以促进初始阶段的脱氢过程, 而酸性基团的存在则是推动后续缩合与环化顺利进行的关键所在; 当催化剂中咪唑的C2位被屏蔽时, 所得催化剂Cat-9的活性出现明显降低, 得到71%的产物收率(Cat-8为88%), 证明了氢键与酸性的协同效应对环化、亚胺的形成以及C—C键的断裂都具有促进作用(Scheme 8b). 最后, 我们以4-硝基苯胺为指示剂测试了离子液体的紫外吸收光谱(Figure 2), 并通过Hammett方程对催化剂的酸度(H0)进行了计算(Table 2)[20]. 结果表明, Cat-8的酸度最强, 可促进酯基水解与脱羧; 而在弱酸存在的条件下, C—C键的断裂受到抑制(Table 2, Entries 2~5, 7); 适宜的酸度是反应物经缩合环化形成二氢喹喔啉的必要条件, 而离子液体的酸度差异是导致产物多样性的主要原因.
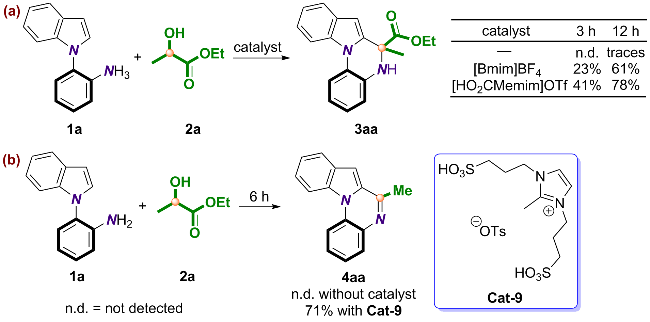
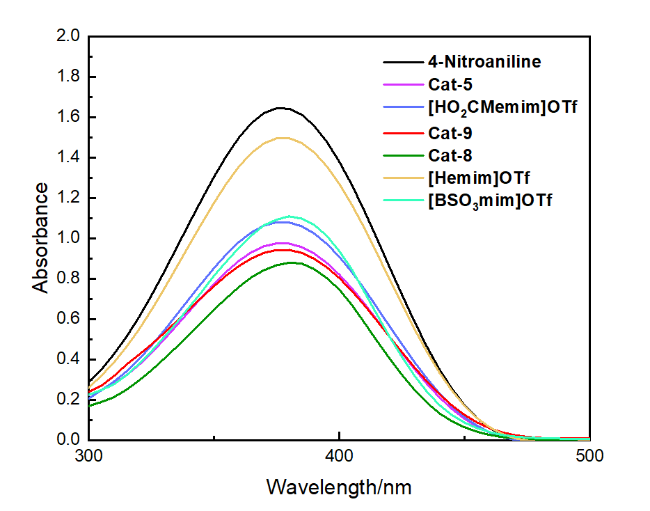
图2 在4-硝基苯胺中测定的离子液体的紫外-可见光谱Figure 2 UV-visible spectra of ILs recorded in 4-nitroaniline |
表2 离子液体的Hammett酸度Table 2 Hammett acidity of ILs |
| Entry | Sample | Amax | [I]/% | [IH+]/% | [I]/[IH+] | H0 |
|---|---|---|---|---|---|---|
| 1 | 4-Nitroaniline | 1.643 | 100 | 0 | — | — |
| 2 | Cat-5 | 0.975 | 59.34 | 40.66 | 1.46 | 1.15 |
| 3 | Cat-8 | 0.875 | 53.25 | 46.75 | 1.14 | 1.05 |
| 4 | Cat-9 | 0.941 | 57.27 | 42.73 | 1.34 | 1.12 |
| 5 | [HO2CMemim]OTf | 1.078 | 65.61 | 34.39 | 1.91 | 1.27 |
| 6 | [Hemim]OTf | 1.459 | 88.80 | 11.20 | 7.93 | 1.89 |
| 7 | [BSO3mim]OTf | 1.087 | 66.15 | 33.85 | 1.95 | 1.28 |
2.6 反应机理的提出
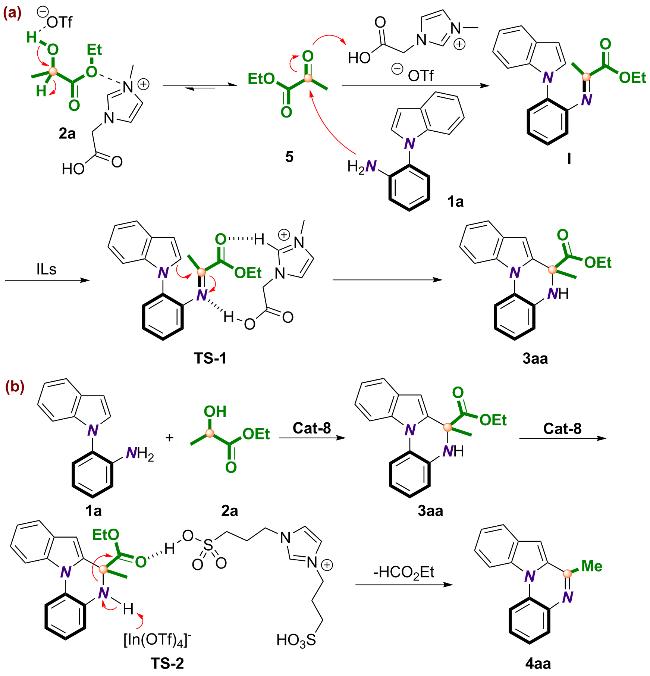
基于上述结果, 如Scheme 9所示, 我们提出脱氢胺化反应机理. 首先是乳酸乙酯的脱氢, 离子液体中的阴离子捕获质子, 而阳离子则通过静电作用夺取氢负离子, 在二者的协同作用下, 乳酸乙酯(2a)经脱氢生成丙酮酸乙酯(5); 接下来是缩合与环化: 丙酮酸乙酯(5)先与2-(1H-吲哚-1-基)苯胺(1a)缩合得到亚胺酯中间体I, 再在离子液体酸性基团与咪唑C2—H氢键的共同作用下经亲电环化得到3aa (I→TS-1→3aa, Scheme 9a); 最后是C—C键的断裂(Cat-8催化体系): 所得中间体3aa经阴离子去质子化, Lewis/Brønsted酸协同活化酯基(3aa→TS-2), 并最终通过C—C键断裂生成4aa (Scheme 9b).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
本工作开发了一种基于α-羟基酯类作为可调C1合成子的绿色合成新策略, 实现了生物质基杂环化合物的差异化合成. 其核心亮点在于化学选择性调控, 即通过可切换的离子液体催化体系精准控制反应路径: 在羧酸型离子液体的催化作用下, 通过脱氢环化高效制备含有季碳中心的环胺类化合物; 而磺酸-Lewis酸双功能型离子液体则适用于合成喹唑啉酮、苯并咪唑、苯并噻唑及喹喔啉等多种氮杂环骨架. 反应机理研究表明, 氢键、静电效应以及酸性基团的协同作用在促进脱羟基胺化、环化和选择性C—C键裂解的串联反应中发挥了多重作用, 催化剂的酸度差异是产物多样化的关键因素. 该方法略整合了可再生原料、绿色可循环催化体系、无金属脱氢及路径的可调性等特点, 具有显著的绿色化学优势, 突显了其在可持续合成领域的重要价值.
(Cheng, B.)