

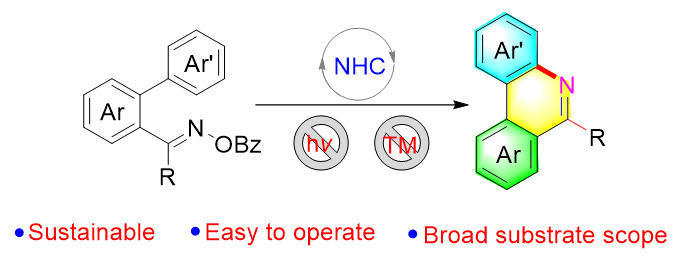
1 引言
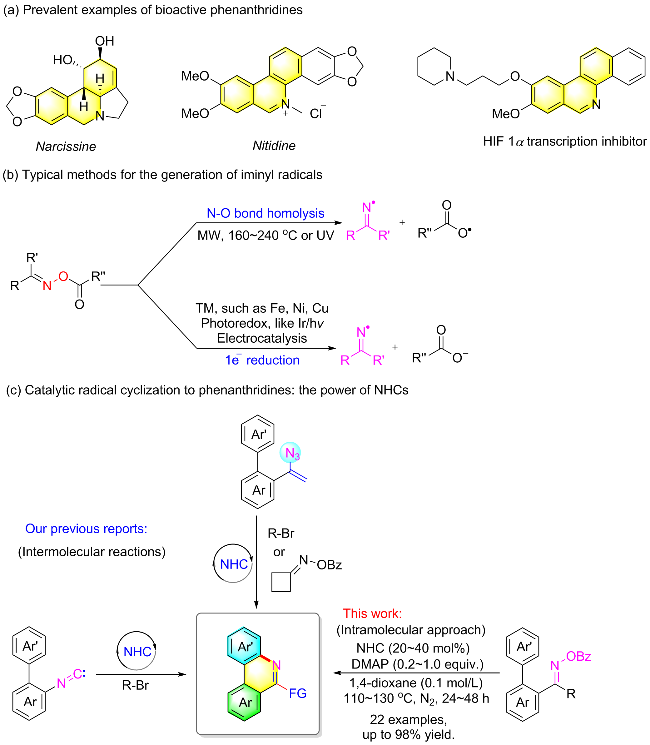
传统上, 亚胺自由基主要通过微波加热(高达240 ℃)[4]或紫外线照射[5](图1b)等苛刻条件使酰基肟发生N—O键均裂而获得. 另外, 酰基肟的N—O键还可以通过单电子还原生成亚胺自由基和羧酸阴离子(图1b)[6-8]. 尽管该领域近期已取得了众多突破, 但许多转化仍高度依赖于过渡金属, 这可能导致药物合成应用中出现金属残留问题[6]. 此外, 这些转化通常还需要外在昂贵的光敏剂催化[7], 如吡啶基-铱络合物和人工合成的光敏剂. 这些局限性导致相关方法在合成通用性、成本效益和环境可持续性方面表现不尽人意. 因此, 迫切需要开发对环境友好、更加经济性的方法: 使用低成本和绿色的催化剂来产生亚胺自由基.
我们课题组[9]一直致力于自由基化学和杂环构建方面的研究. 最近, 我们报道了几种无金属参与、环境友好的分子间自由基加成引发的均裂芳香取代反应(Homolytic aromatic substitution, HAS), 这些反应使用N-杂环卡宾(N-heterocyclic carbene, NHC)催化, 利用乙烯基叠氮[10]或联芳基异腈[11]为原料可快速构建官能化的菲啶(图1c). 这些有趣的结果促使我们进一步探索通过更为直接的分子内反应——直接从酰基肟构建菲啶骨架的可能性. 在本工作中, 我们报道了一种简便且可持续的方法: 通过NHC催化的分子内HAS型自由基环化, 能够快速构建结构多样的众多菲啶骨架(图1c).
2 结果与讨论
2.1 反应条件优化
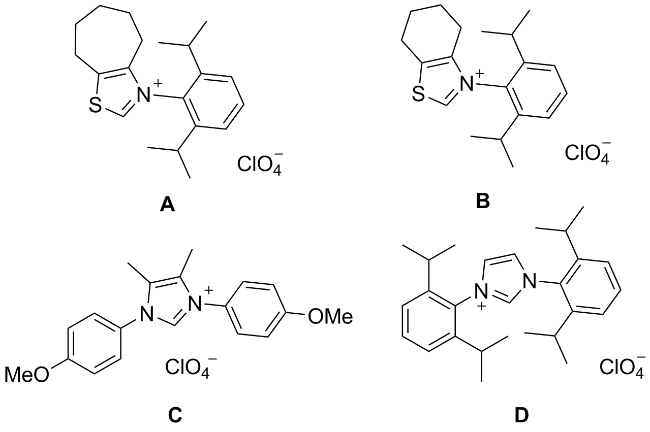
我们以苯甲酰肟1a为底物, 对模板反应展开研究. 经过一定优化后, 在NHC催化下目标产物6-甲基菲啶2a可以88%的核磁产率获得(表1, Entry 1). 表1总结了条件优化过程中的一些代表性结果. 值得注意的是, 其它溶剂(表1, Entries 2~4), 包括乙腈、苯甲醚和二甲基亚砜等, 反应效果均不如1,4-二氧六环. 在所筛选的各种有机碱和无机碱中(表1, Entries 1和5~7), 4-二甲氨基吡啶(4-Dimethylaminopyridine, DMAP)反应效果最好. 其它NHC催化剂(NHC B~D)虽然也可促进该转化, 但效率普遍低于NHC A(图2). 将DMAP的量减少(n碱/ n底物=20%), 反应产率可进一步提高至90%(表1, Entry 11). 然而, 将反应时间缩短至8 h(表1, Entry 12), 导致产物2a的产率显著降低, 因原料1a大部分未转化. 最后, 对照实验证实: NHC催化剂对于该自由基过程顺利进行具有不可或缺的作用(表1, Entry 13).
表1 反应条件优化Table 1 Reaction conditions optimization |
| Entry | Variations | Yielda/% |
|---|---|---|
| 1 | None | 88 |
| 2 | MeCN as the solvent | 70 |
| 3 | PhOMe as the solvent | 35 |
| 4 | DMSO as the solvent | 76 |
| 5 | K3PO4 as the base | 19 |
| 6 | tBuONa as the base | 25 |
| 7 | DBU as the base | 58 |
| 8 | NHC B as the catalyst | 76 |
| 9 | NHC C as the catalyst | 22 |
| 10 | NHC D as the catalyst | 57 |
| 11 | 0.2 equiv. of DMAP | 90 |
| 12b | React for 8 h | 40 |
| 13 | Without NHC A | N.D. |
a Reaction conditions: 1a (0.1 mmol), NHC A (0.02 mmol), DMAP (0.1 mmol), 1,4-dioxane (1.0 mL), 110 ℃, N2, 24 h, NMR yield; b DMAP (0.02 mmol); N.D.: Not detected. DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene. |
2.2 反应底物普适性的考察
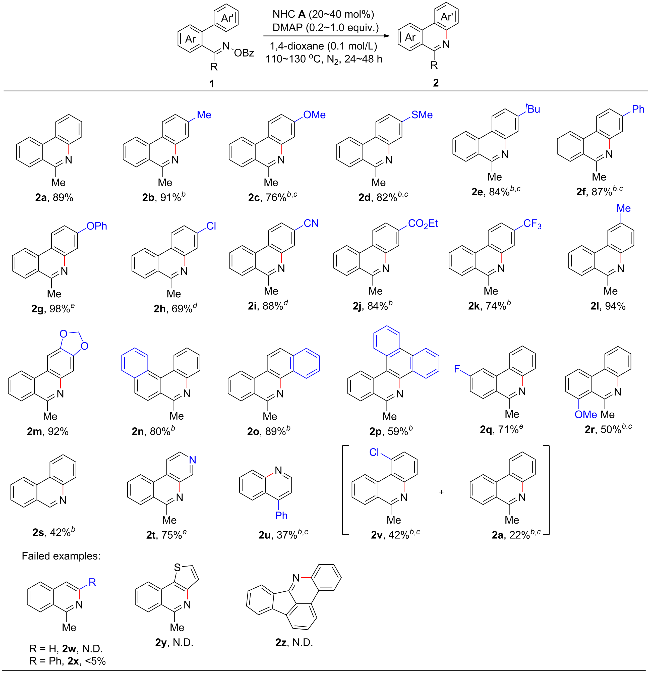
在最佳条件下, 我们进一步研究了亚胺自由基环化反应的底物适用范围(图3). 该转化过程可与多种官能团兼容. 具体来说, 无论取代基的电子性质如何, 如带有甲基、甲氧基、硫甲基、叔丁基、苯基、氯、氰基、三氟甲基和酯基等多种取代基的对位取代苯甲酰肟都能有效地转化为相应的菲啶产物(2a~2k). 另外, 在自由基串联转化过程中, 芳香环上非邻位的卤素也可以很好地兼容(2h和2q). 然而, 当氯原子位于邻位时, 获得了两种不同的产物(2a和2v). 该反应还适用氟原子以及甲氧基或具有类似结构的底物(2c, 2k, 2m, 2q, 2r)[12], 这些基团广泛存在于众多药物分子或生物活性化合物中. 其中, 2r的产率下降是由于邻位甲氧基的位阻所致. 值得说明的是, 间位取代的底物(1l)在此转化中表现出良好的区域选择性(未分离到其它区域异构体). 此外, 具有π扩展体系的底物(1n~1p)也可顺利地得到所需产物(2n~2p). 对于芳醛所形成的肟酯(1s和1u)在此转化中同样有效(2s和2u), 虽然产率有所下降(可能是由于所形成的亚胺自由基中间体不稳定所致). 最后, 芳香杂环肟酯(1t)也可有效地进行转化, 并以75%的产率得到了所需产物(2t). 然而, 由于中间体的稳定性较差或环化张力较大等因素, 也有失败的例子(如2w~2z).
2.3 合成上的应用
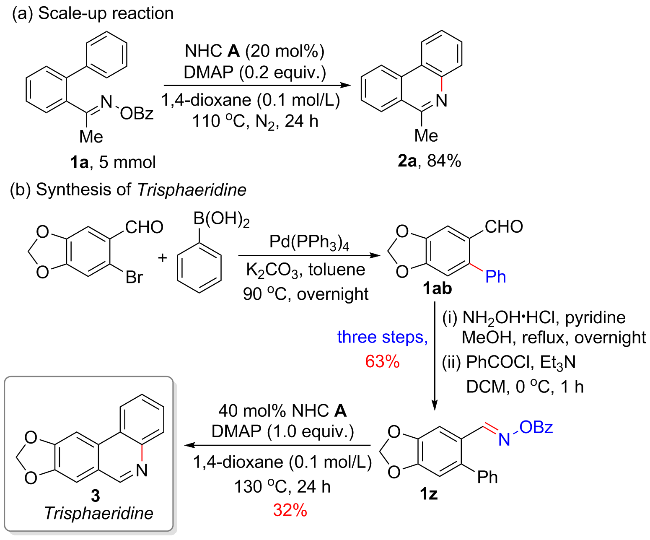
上述转化可进一步放大到克级反应(图4a), 这进一步凸显了这一串联自由基过程的实用性. 此外, 我们将所发展的方法学成功应用到天然生物碱三球啶(Trisphaeridine)的简洁合成中(图4b). 三球啶是从石蒜科开花植物中分离出的一种生物碱, 具有多种药理特性, 包括显著的抗癌活性[13]. 我们的合成策略始于钯催化的Suzuki偶联反应, 以生成相关醛1ab. 随后用盐酸羟胺处理化合物1ab, 再用苯甲酰氯进行酰化, 以良好产率得到酰基肟1z. 最后, 利用所确定的反应条件, 通过1z的自由基环化反应, 以32%的产率获得了生物碱三球啶3, 进一步展现了我们所发展方法在合成复杂分子方面的有效性.
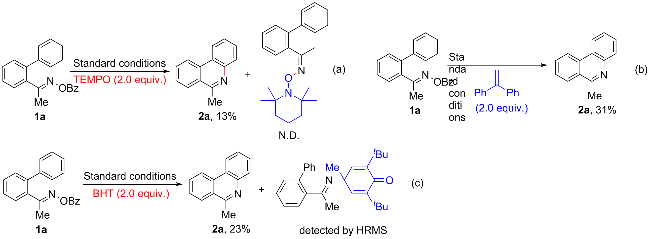
2.4 反应机理研究
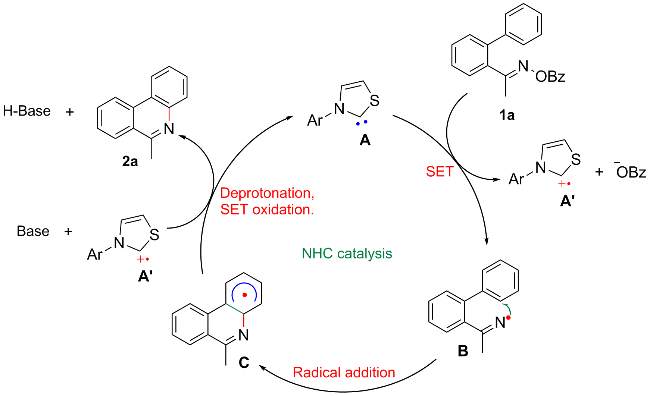
2.5 可能的反应机理
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
综上所述, 我们发展了一种新颖的环境友好型策略: 通过氮杂环卡宾的催化作用, 实现了酰基肟向亚胺自由基的转化, 整个过程无需任何过渡金属、氧化剂或光照. 该方法为高效合成功能化的菲啶类化合物提供了一条便捷途径. 进一步的机理研究表明该反应涉及单电子转移自由基过程. 目前, 我们正在深入探索该转化反应的实用性, 并对其作用机制进行更详细的研究, 相关成果将在后续报道中呈现.
4 实验部分
4.1 NHC催化菲啶合成反应的实验步骤
在氮气氛下, 将酰基肟1 (0.2 mmol, 1.0 equiv.)、氮杂环卡宾A (0.04 mmol, 20 mol%)及4-二甲氨基吡啶(0.04 mmol, 0.2 equiv.)精确称量至Schlenk管中. 随后通过侧管, 利用注射器加入1,4-二氧六环(2.0 mL), 于110 ℃持续搅拌反应24 h. 反应结束后, 待混合物冷却至室温, 加入去离子水(10 mL), 并采用乙酸乙酯(10 mL×3)萃取. 收集有机相后以饱和食盐水(10 mL)洗涤, 经无水硫酸钠干燥, 减压浓缩去除溶剂, 所得残留物利用硅胶柱层析纯化, 经石油醚/乙酸乙酯洗脱, 得产物2.
(Cheng, B.)