

1 引言
共价有机框架(COFs)是一类由C、H、O、N、S等轻质元素组成, 通过共价键连接而成的新型晶态有机多孔聚合物, 具有多样化的构筑基块、种类丰富的成键类型、与“乐高”积木游戏相媲美的高度可控的结构定制设计、可功能化的分子结构、可调谐的光电特性等, 已被证明是用于光催化制备H2O2的热门候选材料[4]. 当前, 通过Schiff-base缩合反应构筑动态可逆亚胺连接COFs被广泛研究[5]. 虽然动态可逆反应可以在合成过程中进行错误检查和自我校正, 在一定程度上可以提升结晶性能, 以便形成长程有序的亚胺COFs, 但亚胺COFs终究是由动态可逆共价键连接, 在光催化反应过程中层状结构容易破坏, 普遍存在化学稳定性较差的缺点, 实际应用受到限制[6]. 此外, 由于亚胺键本征的内在高极化特征的存在[7], 不利于构筑平面内π共轭的COFs, 导致其连接的结构单元之间π电子离域的效率较低, 进而导致分子内电荷转移低、光催化效率较差[8].
近年来, 已经设计了一些策略来提高亚胺COFs的稳定性. 例如, 可通过将亚胺连接COF-TPT-TPA替换为偶氮苯桥联COF-TPT-Azo[9]、将亚胺连接TTA-COF转换为仲胺连接TTA-COF-AR和偶氮连接TTA-Azo-COF[10]、采用合成后替换的方法将亚胺ILCOF-1转化为噻唑键TZ-COF、噁唑键OZ-COF和咪唑键IZ-COF[11]、通过后氧化环化将可逆亚胺连接I-COF转化为不可逆苯并噁唑连接BO-COF[12]、通过aza-Diels-Alder环加成反应将亚胺TAPT-OMe-COF转化为TAPT-OMe-alkyne-COF[13]等, 以实现亚胺COFs稳定性的提升. 或利用分子内氢键[14]、层间氢键[15]、分子对接[16]、结构共振[17]、部分氟化[6]等改善二维亚胺COFs的层间π-π相互作用, 构筑稳定的多层亚胺COFs. 但这些方法只是局部削弱了C=N键的内在动态性质, 并未彻底解决C=N键COFs平面内π-共轭增强的问题. 与传统的动态共价亚胺键相比, 通过不可逆Knoevenagel缩合反应制备而得的sp2-碳共轭共价有机框架获益于稳定的C=C双键连接, 具有卓越的化学稳定性及π共轭特性, 能有效提升π电子在整个骨架中的离域效应并实现高的电子传输效率, 可构筑高效的异相光催化剂, 因而有利于拓宽其应用范围[18]. 然而, 由于C=C成键化学的低可逆性导致全共轭sp2-碳连接COFs的合成仍具有挑战性[19].
作为一种经典的N-杂环共轭化合物, 联吡啶(Bpy)作为光催化氧还原反应(ORR)的活性位点, 可提升COFs光催化产H2O2的性能[20]. 跟其它光催化剂一样, COFs也存在光生载流子分离效率低下的通病, 构筑电子给体-受体(D-A)型COFs是调控光生载流子分离和输运性能的有效策略[21], 其关键在于D-A有效配对. 鉴于Bpy单元本身又具有缺电子的结构特性, 可充当电子受体[22], 因此, 课题组选定2,2'-([2'-联吡啶]-5,5'-二基)二乙腈(Bpy-2CN)连接体扮演电子受体“角色”. 与之同时, 课题组前期研究发现, 具有C3h对称性的平面共轭系统和富含硫的苯并[1,2-b:3,4-b':5,6-b'']三噻吩-2,5,8-三醛(Btt)具有优异的化学稳定性和光电化学活性, 且该单元内部的3个噻吩环混合在苯环中心, 可有效地促进π电子的共轭, Btt与电子受体2,2'-(苯并[c][1,2,5]噻二唑- 4,7-二基双(4,1-亚苯基))二乙腈(Bdd)构筑D-A型Btt-Bdd-COF, 显著增强光催化有机转化性能[23]. 基于此, 有效应用“三合一”集成策略, 以富电子Btt为电子供体, 与强电子受体和ORR活性中心Bpy有效配对, 构筑sp2-碳共轭D-A结构BBpy-COF, 在无牺牲剂体系中开展光催化纯水和氧气制备H2O2的研究. 为进行对比研究, 又分别以Btt和联苯(Bph)为电子供体和弱电子受体[24], 制备对比材料BBph-COF. 本项工作将为合理设计和制备高活性和稳定性的sp2-碳共轭D-A结构COFs提供借鉴和参考.
2 结果与讨论
2.1 合成及结构表征
利用电子给体苯并[1,2-b:3,4-b':5,6-b'']三噻吩-2,5,8-三醛(Btt)分别与电子受体2,2'-([2'-联吡啶]-5,5'-二基)二乙腈(Bpy-2CN)和4,4'-联苯二乙腈(Bph-2CN), 在溶剂热条件下, 通过Knoevenagel缩合反应成功制备了两种sp2-碳连接具有不同“电子推-拉效应”的共价有机框架(图式1), 分别命名为: BBpy-COF和BBph-COF.

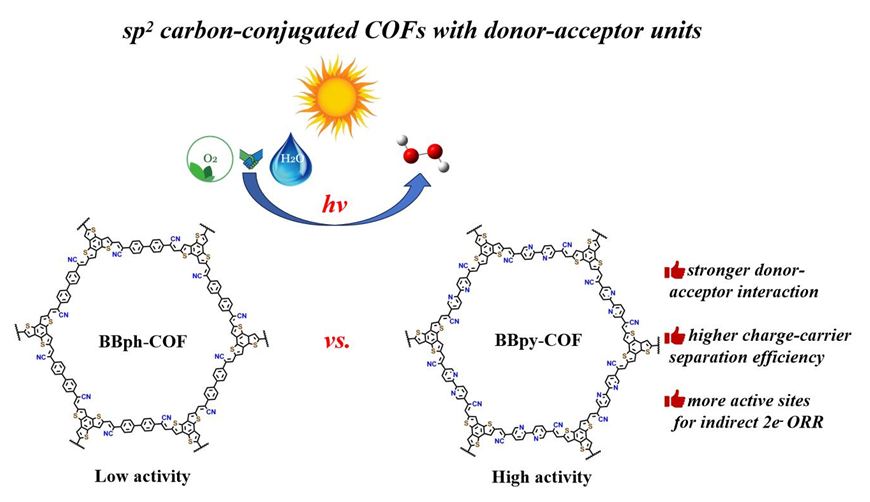
通过粉末X射线衍射(PXRD)对BBpy-COF和BBph-COF的晶体结构进行了表征. 由图1a可见, BBpy-COF的PXRD分别位于4.8°和9.6°的衍射峰与AA堆叠模式模拟的PXRD图谱高度一致, 其中25.8°处的宽峰源于分子结构中部分区域的π-π堆叠作用(见支持信息表S1)[25]. 图1d显示BBph-COF的4.8°和9.7°两个PXRD衍射峰与AA堆叠模式模拟的PXRD图谱吻合度高(见支持信息表S2). 利用傅里叶变换红外光谱(FT-IR)揭示了BBpy-COF和BBph-COF的成键特征. BBpy-COF和BBph-COF分别在2213 cm−1和2211 cm−1处观察到氰基(-CN)信号(图1b和1e)[26], BBpy-COF和BBph-COF在1660 cm−1处归属于醛基团的C=O拉伸振动强度均明显减弱[27], 证明了Knoevenagel缩合反应成功发生. 为进一步佐证BBpy-COF和BBph-COF的成功合成, 利用固体13C交叉极化魔角旋转核磁共振(13C NMR)进行了进一步的表征和分析. 如图1c所示, BBpy-COF的固体13C NMR谱图在δ 100~160范围内具有一组归属于-CN、吡啶环和C=C的特征信号, 且在BBph-COF的固体13C NMR谱中(图1f), -CN与C=C信号峰同样在δ 100~160范围内清晰可见, 进一步证明了BBpy-COF和BBph-COF的成功合成[28-29].
图1 BBpy-COF (a)和BBph-COF (d)的PXRD图谱; Btt, Bpy-2CN, BBpy-COF (b)和Btt, Bph-2CN, BBph-COF (e)的FT-IR谱图; BBpy-COF (c)和BBph-COF (f)的固体13C NMR谱图Figure 1 The PXRD patterns of BBpy-COF (a) and BBph-COF (d); FT-IR spectra of Btt, Bpy-2CN, BBpy-COF (b) and Btt, Bph-2CN, BBph-COF (e); solid-state 13C NMR spectra of BBpy-COF (c) and BBph-COF (f) |
通过扫描电子显微镜(SEM)和透射电子显微镜(TEM)对BBpy-COF和BBph-COF的形貌进行了表征. BBpy-COF的SEM图像呈花球形(图3a), 而BBph-COF的SEM图像则表现出条状形貌(图3b), 二者的TEM图像均显示为明显的层状结构(图3c和3d). 此外, 利用能谱仪(EDS)探测了BBpy-COF和BBph-COF的元素分布. BBpy-COF和BBph-COF的EDS元素映射分析均清楚显示C、N和S元素的均匀分布(见支持信息图S2). 为了探究BBpy-COF和BBph-COF的比表面积和孔隙率特征, 在77 K条件下对其进行了N2吸附-脱附测试(见支持信息图S3). 通过Brunauer-Emmett-Teller (BET)方程计算所得到的BBpy-COF和BBph-COF的比表面积分别为78.9 m2•g−1和78.1 m2•g−1. 利用非局域密度泛函理论(NLDFT)模型计算得到BBpy-COF和BBph-COF均为介孔结构. 最后, 为了研究两种COFs的热稳定性, 在N2气氛下对二者进行了热重测试(TGA). 虽然BBpy-COF和BBph-COF在300~400 ℃时没有明显的质量损失, 但是BBpy-COF的热稳定性总体要优于BBph-COF (见支持信息图S4).

2.2 光学和光电化学分析
为了进一步探究BBpy-COF和BBph-COF的光催化性能, 通过紫外-可见(UV-Vis)吸收光谱比较了两者的光吸收能力和光吸收范围的差异. 如图4a所示, BBpy-COF对可见光具有强而宽的吸收, 其吸收带边与BBph-COF相比, 明显出现了79 nm的红移[32]. 利用Kubelka-Munk函数和Tauc图(图4b)计算BBpy-COF和BBph-COF的光学带隙(Eg)[33], BBpy-COF光学带隙 (Eg=2.14 eV)明显窄于BBph-COF (Eg=2.23 eV). 进一步采用密度泛函理论(DFT)计算研究BBpy-COF和BBph-COF的最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)分布特征. 由图4c可知, BBpy-COF的HOMO和LUMO分别主要分布于Btt和Bpy. 与之不同, BBph-COF的HOMO轨道电子主要集中在C=C键、Btt和Bph与C=C键连接的苯环上, 而其LUMO轨道主要局域于C=C键、Btt与C=C键连接的噻吩环和Bph与C=C键连接的苯环上(见支持信息图S5). 上述差异表明, 在光激发过程中BBpy-COF的电子更易从Btt跃迁至Bpy, 即更强的“电子推-拉效应”触发了更强、更宽的可见光吸收, 从而形成了带隙更窄的半导体[34]. 此外, 通过电化学Mott-Schottky (M-S)测试, 可以获取半导体类型和平带电位. BBpy-COF和BBph- COF的M-S曲线均呈现正斜率(见支持信息图S6), 表明两者均为典型的n型半导体[35]. 由于n型半导体的导带底(CBM)比平带电势更负-0.1 V[36], BBpy-COF的CBM拟合为-1.23 V (vs. Ag/AgCl) (对应于-1.03 V (vs. NHE)), BBph-COF的CBM拟合为-1.20 V (vs. Ag/AgCl) (对应于-1.00 V (vs. NHE)). 根据公式Eg=EVB-ECB[26], 可以进一步计算出BBpy-COF和BBph-COF的价带顶(VBM)分别为1.11 V (vs. NHE)和1.23 V (vs. NHE), 即可得到BBpy-COF和BBph-COF相应的能带结构(见支持信息图S7). 从热力学角度分析, BBpy-COF和BBph-COF的CBM均负于O2的两电子还原电势, 表明两者的还原电势对于光催化还原O2生成 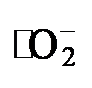 中间体是热力学可行的, 而
中间体是热力学可行的, 而 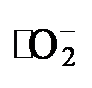 中间体可进一步转化为H2O2[37].
中间体可进一步转化为H2O2[37].
中间体是热力学可行的, 而 中间体可进一步转化为H2O2[37].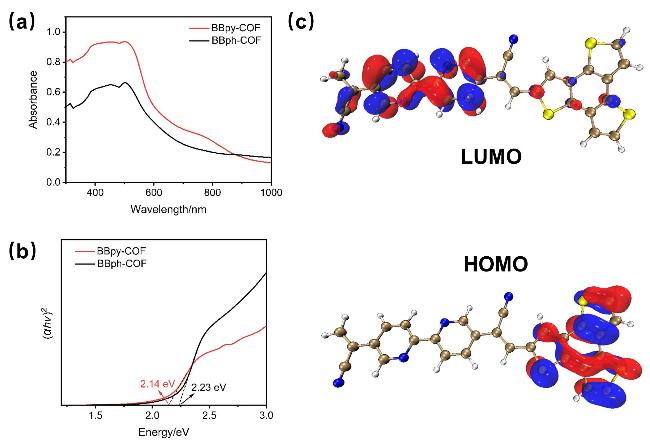
利用CHI 660E电化学工作站对所制备的两种COFs的瞬态光电流响应进行了分析, 以考察光生电子的迁移速率. 如图5a所示, BBpy-COF具有更高的瞬态光电流密度, 意味着更多的光生电子通过外电路从BBpy-COF迁移到对电极[38]. 图5b展示了BBpy-COF和BBph-COF的电化学阻抗谱(EIS), BBpy-COF在光催化剂/电解液界面处表现出较低的电荷转移电阻(Rct), 表明BBpy-COF的光生电子扩散迁移率比BBph-COF大[39]. 此外, 由图5c可知, BBpy-COF的光致发光(PL)光谱强度与BBph-COF相比明显减弱, 这表明BBpy-COF有效抑制了光生电子-空穴对的重组, 并推动光生电子向催化活性位点转移/迁移, 进而促进后续光催化产H2O2[40]. 从图5d可计算出BBpy-COF和BBph-COF的平均荧光寿命分别为1.12 ns和0.87 ns. BBpy-COF更长的荧光预期寿命意味着其具有较长的光生载流子扩散时间, 延长的载流子寿命有利于光催化产H2O2[41]. 综合以上分析, 与BBph-COF相比, BBpy-COF能够加速光生电子-空穴对的分离和迁移, 能更有效抑制其复合.

2.3 光催化产双氧水性能及其机理
在不添加任何牺牲剂的情况下, 对BBpy-COF和BBph-COF在O2饱和纯水中光催化产H2O2性能进行了评价. 采用草酸钛钾法测定H2O2含量(见支持信息图S8). 由图6a可知, 随着可见光辐照时间的延长, BBpy-COF和BBph-COF的H2O2产量逐渐增加. 与BBph-COF相比, BBpy-COF光催化产H2O2在3 h内呈现出更好的线性增长, 表明其光催化产H2O2是一个稳定的连续过程. 此外, BBpy-COF具有更高的光催化H2O2生成速率, 达到1394.7 µmol•g−1•h−1, 比BBph-COF (656.5 µmol•g−1•h−1)提高了2.12倍(见支持信息图S9). 连续五次光催化循环实验后(图6b), BBpy-COF未见明显失活, 且其PXRD和FT-IR光谱都没有明显变化(见支持信息图S10). 因此, 所制备的BBpy-COF在光催化合成H2O2过程中表现出良好的循环稳定性.

图6 BBpy-COF和BBph-COF光催化产H2O2的动力学分析(a); BBpy-COF光催化产H2O2循环实验(b); 不同实验条件下BBpy-COF光催化产H2O2 (c); BBpy-COF自由基淬灭实验(d); BBpy-COF的DMPO- |

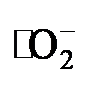
为了研究BBpy-COF光催化合成H2O2机理, 实施了不同条件下的对照实验和自由基淬灭实验. 由图6c可知, 在黑暗条件下或不添加任何光催化剂时, 几乎都没有检测到H2O2, 这表明可见光和光催化剂对光催化产H2O2都是必不可少的. 在空气条件下进行光催化产H2O2, BBpy-COF光催化性能明显下降; 当O2被氩气(Ar)取代, 几乎检测不到H2O2, 表明富氧气氛对光催化产H2O2至关重要, 且在可见光照射下BBpy-COF主要通过还原O2生成H2O2[42]. 此外, 叔丁醇(t-BA)、C2H5OH、AgNO3和对苯醌(p-BQ)分别用作•OH、h+、e−和 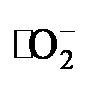 的捕获剂(图6d). 引入t-BA对光催化产H2O2影响较小, 说明•OH不是光催化ORR的中间体. 然而, 通过添加C2H5OH来捕获空穴, 其为光催化ORR提供了更多的光生电子, 进而提高光合成H2O2的生成速率. 值得注意的是, 在AgNO3或p-BQ存在下, 光催化合成H2O2生成速率被显著抑制, 这表明光催化产H2O2的主要活性种是e−和
的捕获剂(图6d). 引入t-BA对光催化产H2O2影响较小, 说明•OH不是光催化ORR的中间体. 然而, 通过添加C2H5OH来捕获空穴, 其为光催化ORR提供了更多的光生电子, 进而提高光合成H2O2的生成速率. 值得注意的是, 在AgNO3或p-BQ存在下, 光催化合成H2O2生成速率被显著抑制, 这表明光催化产H2O2的主要活性种是e−和 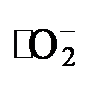 . 简言之, BBpy-COF自由基淬灭实验证明了光催化ORR为两步单电子路线(O2还原为
. 简言之, BBpy-COF自由基淬灭实验证明了光催化ORR为两步单电子路线(O2还原为 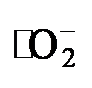 , 再还原为H2O2)[24].
, 再还原为H2O2)[24].
的捕获剂(图6d). 引入t-BA对光催化产H2O2影响较小, 说明•OH不是光催化ORR的中间体. 然而, 通过添加C2H5OH来捕获空穴, 其为光催化ORR提供了更多的光生电子, 进而提高光合成H2O2的生成速率. 值得注意的是, 在AgNO3或p-BQ存在下, 光催化合成H2O2生成速率被显著抑制, 这表明光催化产H2O2的主要活性种是e−和 . 简言之, BBpy-COF自由基淬灭实验证明了光催化ORR为两步单电子路线(O2还原为 , 再还原为H2O2)[24].为了进一步确认光催化产H2O2的途径, 以5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)为探针进行了电子顺磁共振(EPR)测试和电化学旋转圆盘电极(RDE)测定. 在图6e所示的EPR光谱中, BBpy-COF在黑暗条件下没有检测到 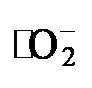 信号, 而在光照射5 min后, 则检测到DMPO-
信号, 而在光照射5 min后, 则检测到DMPO- 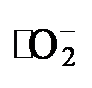 的强烈共振信号, 表明在光催化合成H2O2过程中产生了更多的
的强烈共振信号, 表明在光催化合成H2O2过程中产生了更多的 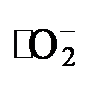 自由基[43]. 此外, 由图6f可见(见支持信息图S11), 通过Koutecky-Levich方法拟合曲线计算出BBpy-COF的平均转移电子数(n)为2.27, 其接近两电子ORR的理论值(n=2), 进一步证明了BBpy-COF在可见光照射下主要通过两电子ORR途径生成H2O2[44]. 综上所述, 结合能带结构、不同条件下对照实验、自由基淬灭实验、EPR光谱和ORR的n值测定, 确认了BBpy-COF在可见光照射下由纯水和氧气通过两步单电子ORR生成H2O2[45].
自由基[43]. 此外, 由图6f可见(见支持信息图S11), 通过Koutecky-Levich方法拟合曲线计算出BBpy-COF的平均转移电子数(n)为2.27, 其接近两电子ORR的理论值(n=2), 进一步证明了BBpy-COF在可见光照射下主要通过两电子ORR途径生成H2O2[44]. 综上所述, 结合能带结构、不同条件下对照实验、自由基淬灭实验、EPR光谱和ORR的n值测定, 确认了BBpy-COF在可见光照射下由纯水和氧气通过两步单电子ORR生成H2O2[45].
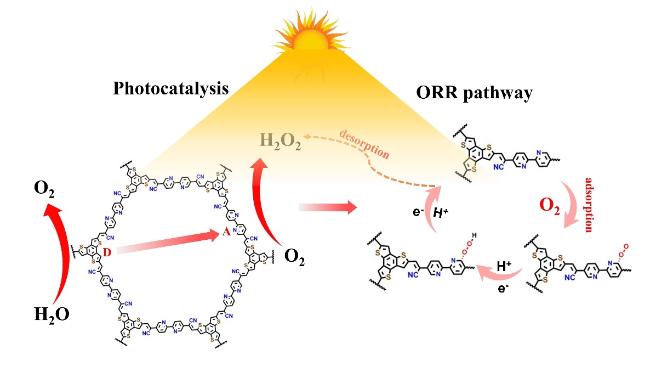
信号, 而在光照射5 min后, 则检测到DMPO- 的强烈共振信号, 表明在光催化合成H2O2过程中产生了更多的 自由基[43]. 此外, 由图6f可见(见支持信息图S11), 通过Koutecky-Levich方法拟合曲线计算出BBpy-COF的平均转移电子数(n)为2.27, 其接近两电子ORR的理论值(n=2), 进一步证明了BBpy-COF在可见光照射下主要通过两电子ORR途径生成H2O2[44]. 综上所述, 结合能带结构、不同条件下对照实验、自由基淬灭实验、EPR光谱和ORR的n值测定, 确认了BBpy-COF在可见光照射下由纯水和氧气通过两步单电子ORR生成H2O2[45].通过以上一系列实验表征、DFT计算和借鉴相关参考文献, 阐述BBpy-COF光催化产H2O2机理. 由图7可知, 在可见光辐照下, 由于BBpy-COF具有更强和更宽的可见光吸收, 产生了更多的光生电子-空穴对. 鉴于电子给体Btt和电子受体Bpy构建了强D-A结构且两者通过C=C双键连接提升框架共轭度, 产生了更强的“电子推-拉效应”, 提升了光生电子-空穴对的分离与迁移能力. 一方面, 光生空穴富集在Btt, 随即被4h+水氧化所消耗; 另一方面, 光生电子则富集于ORR活性位点Bpy, 通过两步单电子ORR路径生成H2O2.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
通过一步溶剂热法, 采用Knoevenagel缩合反应成功构筑了两种基于sp2-碳连接具有不同“电子推-拉效应”的共价有机框架, 并对其进行光催化产双氧水性能研究. BBpy-COF表现出更佳的光催化产H2O2性能, 其生成速率可达1394.7 µmol•g−1•h−1, 为BBph-COF (656.5 µmol•g−1•h−1)的2.12倍. 借助一系列实验表征和DFT计算, 阐明相应光催化产H2O2机理. BBpy-COF凭借电子给体Btt和电子受体Bpy构筑强D-A结构且通过共轭C=C双键连接, 产生了更强的D-A作用, 表现为增强的可见光吸收能力、更好的光生载流子分离与迁移能力, 依靠ORR活性位点Bpy, 高效通过两步单电子ORR路径实现光催化产H2O2. 本项工作为合理设计和构筑高效共价有机框架仅利用自然界阳光、水和O2制备H2O2提供了一定的借鉴和参考.
4 实验部分
4.1 COFs合成
将Btt (0.04 mmol, 13.22 mg)和Bpy-2CN (0.06 mmol, 14.06 mg)添加到10 mL Pyrex管中, 并向其中加入1,2-二氯苯(o-DCB)和正丁醇(n-BuOH) (V/V=2∶1, 2 mL)的混合溶液. 将混合溶液超声处理15 min, 再添加0.2 mL 3 mol/L四丁基氢氧化铵(TBAH)到上述均匀分散的溶液中. 将获得的溶液超声处理2 min, 并用液氮和双排管连续进行至少三次“冷冻-泵-解冻”循环脱气处理. 将Pyrex管真空密封后, 放入电热恒温鼓风干燥箱维持120 ℃反应72 h. 将反应完全的样品冷却至室温后, 离心收集样品, 用四氢呋喃(THF)和丙酮洗涤数次, 直至滤液无色. 该样品进一步用四氢呋喃索氏提取12 h, 随后在60 ℃真空干燥12 h, 得到最终样品, 标记为BBpy-COF. 除了用Bph-2CN (0.06 mmol, 13.94 mg)替换Bpy-2CN (0.06 mmol, 14.06 mg)之外, 保持其它条件不变, 即可制备对比材料BBph-COF.
4.2 光催化产H2O2
称取COFs样品5 mg, 超声(10 min)分散于30 mL去离子水中, 将溶液转移至光反应器中并密封, 在黑暗条件下向体系中通入O2搅拌30 min以达到吸附-解吸平衡. 随后将光反应器放置于PLS-SXE300/300UV型号氙灯光源下反应3 h, 期间保持20 W输出光功率, 不断通入氧气并搅拌. 采用循环冷却水系统, 温度控制在25 ℃. 反应结束后, 用注射器从光反应器中抽取混合溶液, 并利用0.45 µm微孔滤膜过滤得到澄清的H2O2溶液. 取过滤后的H2O2溶液1.5 mL与显色剂1 mL摇匀稳定6 min后在紫外分光光度计下测定400 nm波长所对应的吸光度[46], 利用H2O2浓度-吸光度标准曲线, 即可评价样品光催化产H2O2性能.
(Cheng, B.)