




1 引言
2 不同种类的网络结构设计
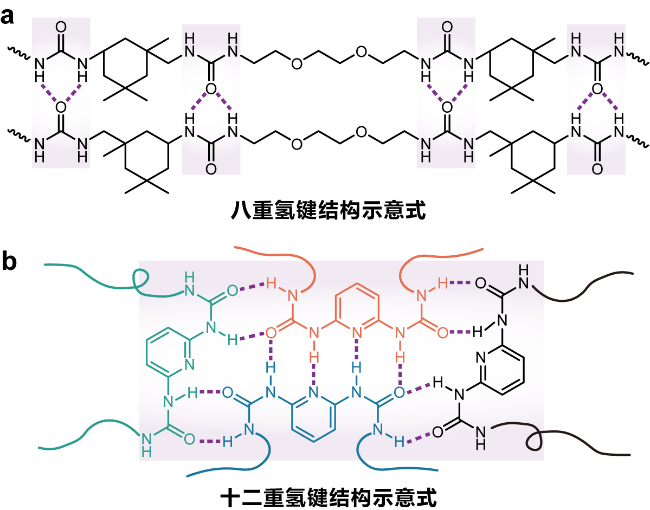
2.1 氢键交联的网络
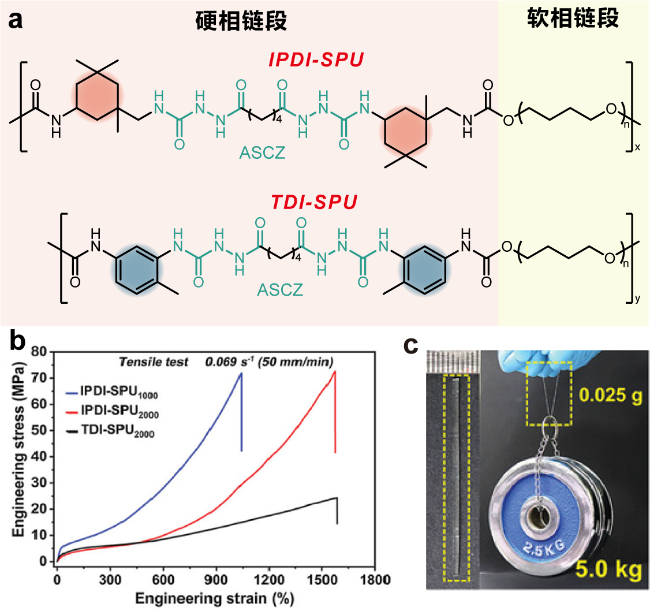
图1 (a) IPDI-SPU和TDI-SPU的结构式; (b) IPDI-SPU和TDI-SPU的应力-应变曲线; (c) IPDI-SPU2000弹性体条(25 mg)可以举起5.0 kg的重量[50]Figure 1 (a) Chemical structures of IPDI-SPU and TDI-SPU elastomers. (b) Stress-strain curves of IPDI-SPU and TDI-SPU elastomers. (c) Photographs showing that the IPDI-SPU2000 elastomer strip (25 mg) can lift a weight of 5.0 kg[50] |
2.2 配位键交联的网络

图3 (a)甲硅烷保护基的裂解和随后铁络合物形成的过程, 以及在此过程中试样膨胀的展示; (b, c)铁离子处理前后的网络结构示意图; (d)不同网络的应力-应变曲线[62]. (e)聚合物主链的结构式与不同离子配位后的网络结构; (f)不同网络的应力-应变曲线; (g)不同网络的杨氏模量和韧性值[63]Figure 3 (a) A scheme of the cleavage of the silyl protective groups and subsequent iron complex formation, and a depiction of the swelling of a test specimen during the process. Schematics of the network structure (b) before and (c) after iron treatment. (d) Stress-strain curves of different networks[62]. (e) Schematics of the polymer backbone and network structures after coordination of different ions. (f) Stress-strain curves, (g) Young's modulus and toughness values of different networks[63] |
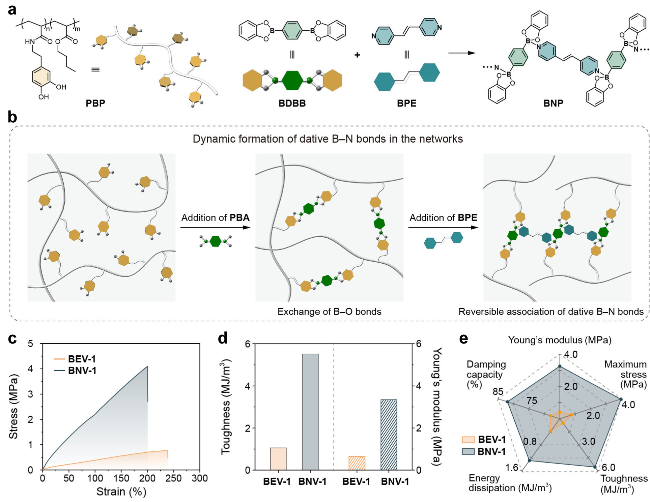
图4 (a) PBP的化学结构和示意图及通过B—N配位键形成的B—N配位超分子聚合物(BNP)的示意图; (b)通过B—O和B—N键形成超分子交联聚合物; 不同网络的(c)应力-应变曲线, (d)杨氏模量和韧性数值以及(e)其他机械性能的对比[66]Figure 4 (a) Chemical structure and cartoon representation of PBP and the dative B—N supramolecular polymer (BNP) via dative B—N bonds. (b) Cartoon representation of the formation of the supramolecular polymer cross-linked vitrimer via dynamic B—O and B—N dative bonds. Comparison of (c) stress-strain curves, (d) Young's modulus and toughness values, (e) and other mechanical properties of different networks[66] |
2.3 主客体分子交联的网络
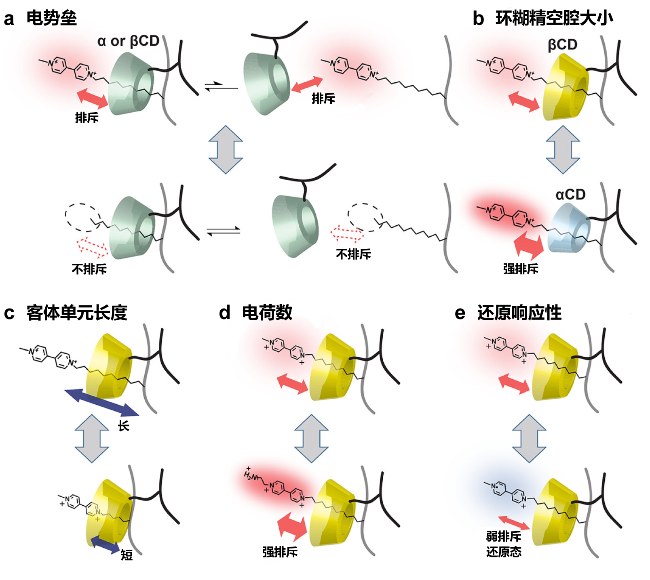
图5 CD基超分子水凝胶的机械性能受CD和聚合物侧链上客体分子的影响: (a)电势垒, (b) CD腔尺寸, (c)客体单元长度, (d)电荷数和(e)还原响应性[73]Figure 5 Mechanical properties of the CD-based supramolecular hydrogels affected by CDs and guest molecule on polymer side chain: (a) electric barrier, (b) CD cavity size, (c) length of guest unit, (d) charge number, and (e) reduction responsiveness[73] |
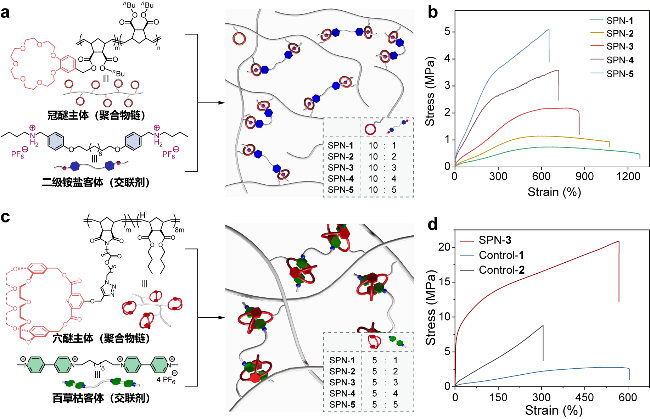
图6 (a)冠醚主体聚合物链、二级铵盐客体交联剂和超分子网络的示意图; (b)不同交联密度超分子网络的应力-应变曲线[74]; (c)穴醚主体聚合物链、百草枯客体交联剂和超分子网络的示意图; (d)不同聚合物网络的应力-应变曲线[76]Figure 6 (a) Chemical structures and schematic representations of crown ether-decorated polymer chain, secondary ammonium salts cross-linker and supramolecular network (SPN). (b) Stress-strain curves of SPNs with different cross-linking densities[74]. (c) Chemical structures and schematic representations of cryptand-decorated polymer chain, bisparaquat cross-linker and SPN. (d) Stress-strain curves of different polymer networks[76] |
2.4 力敏基团增强的聚合物
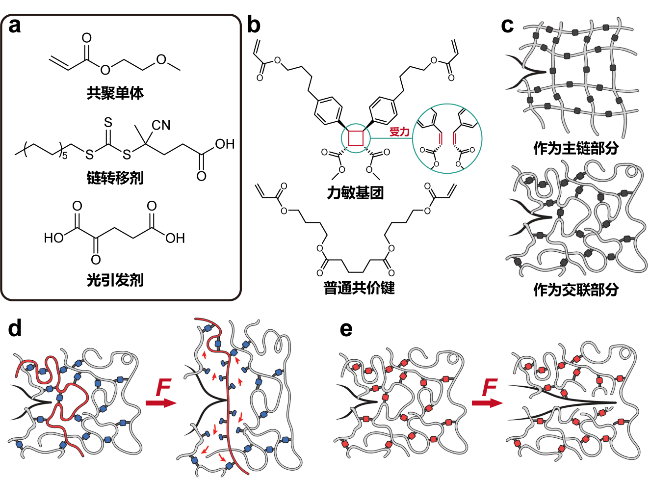
图7 (a)聚合反应所需的分子; (b)力敏基团和普通共价键的结构式; (c)构筑网络的不同方式; (d)力敏基团构筑的网络和(e)普通共价键构筑的网络在外力作用下的断裂示意[80]Figure 7 (a) Molecules required for the polymerization reaction. (b) Structural formulas of force-sensitive group and normal covalent bonds. (c) Different ways of constructing the network. (d) Schematic representation of the fracture of the network constructed by force-sensitive groups and (e) normal covalent bonds upon external force[80] |

图8 (a)初始网络示意图; (b)力敏基团在超声作用下的结构变化; (c)力诱导的网络交联示意图; (d)不同超声处理持续时间后样品的应力-应变曲线; (e)超声前后聚合物的紫外-可见光谱; (f)不同超声处理持续时间后样品的储能模量变化[82]Figure 8 (a) Schematic representation of the initial network. (b) Structural changes of force-sensitive groups under sonicating. (c) Schematic representation of cross-linking behavior induced by force. (d) Stress-strain curves of the samples with different sonicating durations. (e) Ultraviolet-visible spectroscopy of the polymer with and without sonicating. (f) Storage modulus changes of the samples after different sonicating durations[82] |
2.5 机械互锁聚合物网络
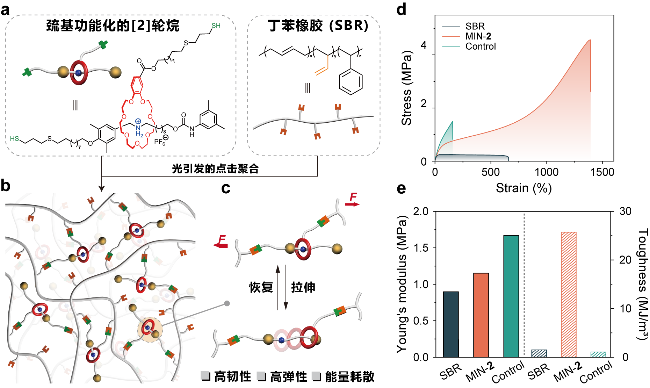
图9 (a)轮烷交联剂和丁苯橡胶(SBR)的化学结构及相应示意图; (b)机械互锁网络(MINs)的示意图; (c)机械互锁交联点在外力作用下的运动行为; SBR、机械互锁网络MIN和对照样品control的(d)应力−应变曲线、(e)杨氏模量和韧性值[99]Figure 9 (a) Chemical structures and corresponding schematic representations of rotaxane cross-linker and styrene-butadiene rubber (SBR). (b) Schematic illustration of the formation of mechanically interlocked networks (MINs). (c) Molecular motion of mechanically interlocked cross-links upon external force. (d) Stress-strain curves, (e) Young’s modulus and toughness of SBR, MIN and control[99] |

图10 (a)由CDs组成的可移动交联点交联的滑环凝胶. PEG的紧密堆积结构在拉伸和释放过程中形成和破坏; (b)不同聚合物网络的应力-应变曲线[101]Figure 10 (a) Slide-ring (SR) gel with movable cross-links composed of CDs. The close-packed structure of PEG is formed and destroyed during stretching and releasing. (b) Stress-strain curves of different polymer networks[101] |
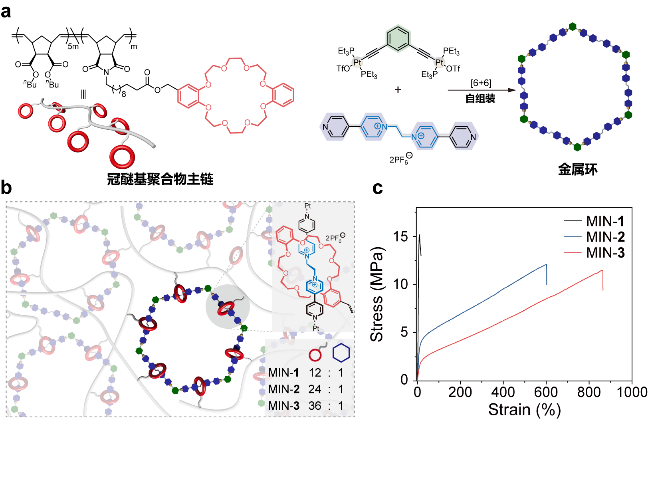
图11 (a)冠醚基聚合物主链、金属环交联剂的化学结构和示意图; (b)分子项链交联形成的机械互锁网络示意图; (c)不同交联密度的聚合物网络的应力-应变曲线[105]Figure 11 (a) Chemical structure and corresponding cartoon of crown ether-based polymer and metallacycle cross-linker. (b) Mechanically interlocked networks (MINs) cross-linked by molecular necklace. (c) Stress-strain curves of polymer networks with different cross-linking densities[105] |
2.6 其他作用改性的聚合物


图13 (a)通过硬−软−硬立体三嵌段共聚物it-MRxM (x=m/n)与st-PMMA的物理交联增韧TPE立体复合物; (b, c) [it-PRMA]/[it-PMMA]=4或6的立体络合全甲基丙烯酸弹性体的应力-应变曲线[117]Figure 13 (a) Toughened TPE stereocomplex via physical cross-linking in a hard-soft-hard stereo-triblock co-polymer, it-MRxM (x=m/n), with st-PMMA. (b, c) Stress-strain curves of stereocomplexed all-methacrylic elastomers with [it-PRMA]/[it-PMMA]=4 or 6[117] |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}