




1 引言
硒(Se)是人体必需的非金属微量元素之一, 在人体的新陈代谢中起着非常重要的作用[1]. 手性含硒有机化合物在合成化学、材料科学、医药及农药等领域具有广泛的应用[2]. 将天然产物[3]、药物分子[4]和材料分子[5]中的氧原子或硫原子替换为同族的硒原子往往能极大地提高其生物活性或材料性能. 同时, 在不对称合成中手性有机硒化合物常被用作手性配体或直接作为手性有机小分子路易斯碱催化剂在多种反应中使用[6]. 因此, 发展高效的手性有机硒化合物合成方法具有重要的研究意义和重大的应用价值. 经典的合成方法大多基于“手性池”(chiral-pool)策略[7], 这些方法以廉价易得的天然产物作为手性原料出发来合成手性有机硒化合物, 但往往存在步骤经济性和原子经济性较低的问题. 相比而言, 不对称催化策略是合成该类化合物最经济的方法之一. 以单质硒为硒源构建C—Se键是合成有机硒化合物最直接的方式[8]. 然而, 硒单质存在反应活性低、在有机溶剂中的溶解性不高以及易于形成过渡金属-硒团簇等问题, 因而从硒粉出发以不对称催化的方式直接合成手性有机硒化合物的方法极具挑战性, 目前尚未见诸文献报道. 近年来, 合成化学家们发展了一系列构建手性有机硒化合物的不对称催化合成方法[9], 主要分为以下两种策略: (1)含硒合成砌块的不对称转化, 反应过程中不涉及C—Se键的形成与断裂[10]; (2)有机硒试剂与相应底物之间的立体选择性硒基化反应, 反应过程中伴随新的C—Se键的形成. 后者是目前构建手性有机硒化合物最为直接有效的方法之一. 目前, 见诸文献报道的硒基化反应的有机硒试剂包括亲电型、亲核型和自由基型三种. 常见的亲电性和亲核性有机硒试剂见图1. 此外, 硒醚也可以作为亲核试剂与亲电底物反应, 生成相应的硒鎓离子中间体. 关于自由基硒基化反应的研究相对较少[11], 目前尚没有关于有机硒试剂与相应底物的不对称自由基反应方面的报道. 本篇综述根据硒化试剂种类的不同进行分类, 总结了近十年来(2016~至今)通过有机硒试剂与相应底物的催化不对称反应制备手性有机硒化合物的研究进展, 并对一些代表性方法学的反应机理和底物适用范围进行了详细介绍, 最后对该领域的未来发展进行了展望.
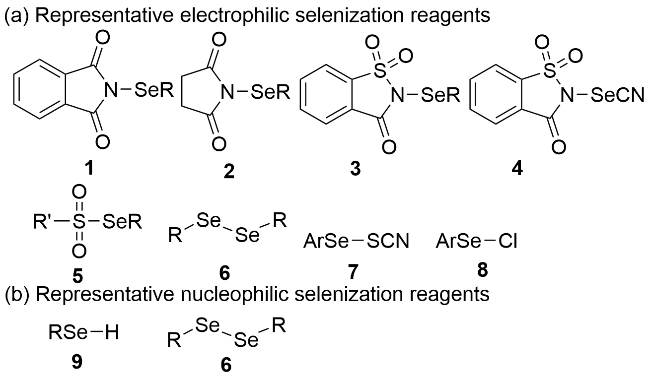
2 亲电有机硒试剂与亲核底物的立体选择性反应
与亲电有机硒试剂反应的亲核试剂(或亲核中间体)包括烯烃、芳烃和烯醇化合物(Scheme 1). 在手性催化剂的活化和诱导下, 亲电硒源立体选择性地进攻烯烃的π键形成具有特定立体构型的三元环状硒鎓离子中间体, 随后发生亲核开环反应生成最终的双官能团化的手性产物. 若亲核试剂与烯烃在同一分子上, 则生成环状手性产物. 当α,β-不饱和醇用作亲核底物时则有可能发生半频哪醇重排反应. 通过亲电硒源与联芳基化合物的邻位C(sp2)—H键阻转选择性硒基化反应是高效构建轴手性有机硒化合物的一种非常重要的策略. 烯醇物种的对映选择性硒基化反应也是合成手性含硒有机分子的一种常用策略. 在碱性条件下夺取羰基化合物的α-氢是生成烯醇物种最直接的方式. 此外, α,β-不饱和羰基化合物经Michael加成反应、α-重氮酯与亲核试剂的反应也可以生成烯醇化物中间体. 本节根据底物结构和反应类型的不同, 从烯烃的立体选择性反应、芳烃的C(sp2)—H键阻转选择性硒基化反应和烯醇化合物的对映选择性α-硒基化反应等三方面展开叙述.
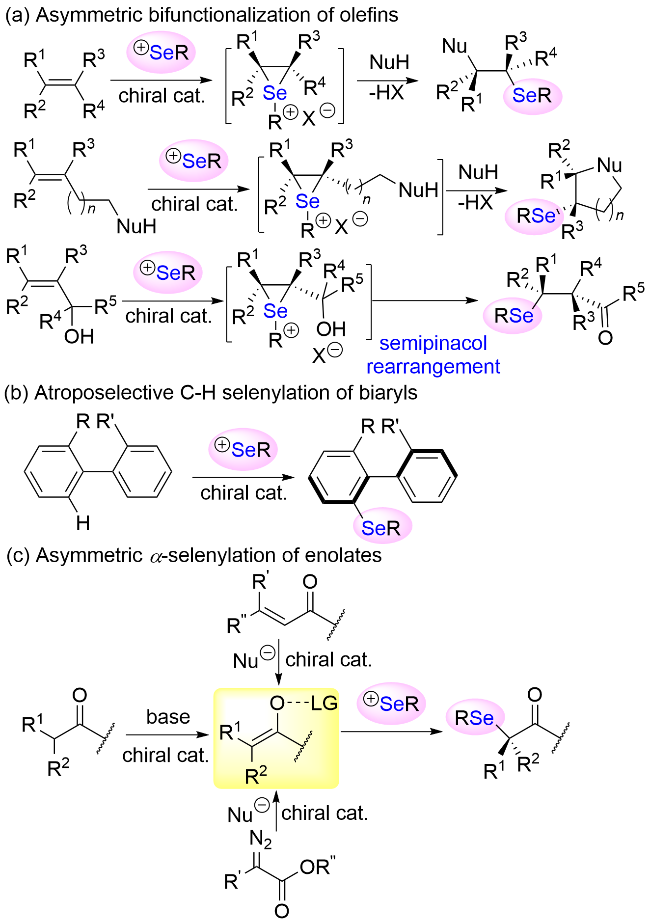
2.1 烯烃的立体选择性反应
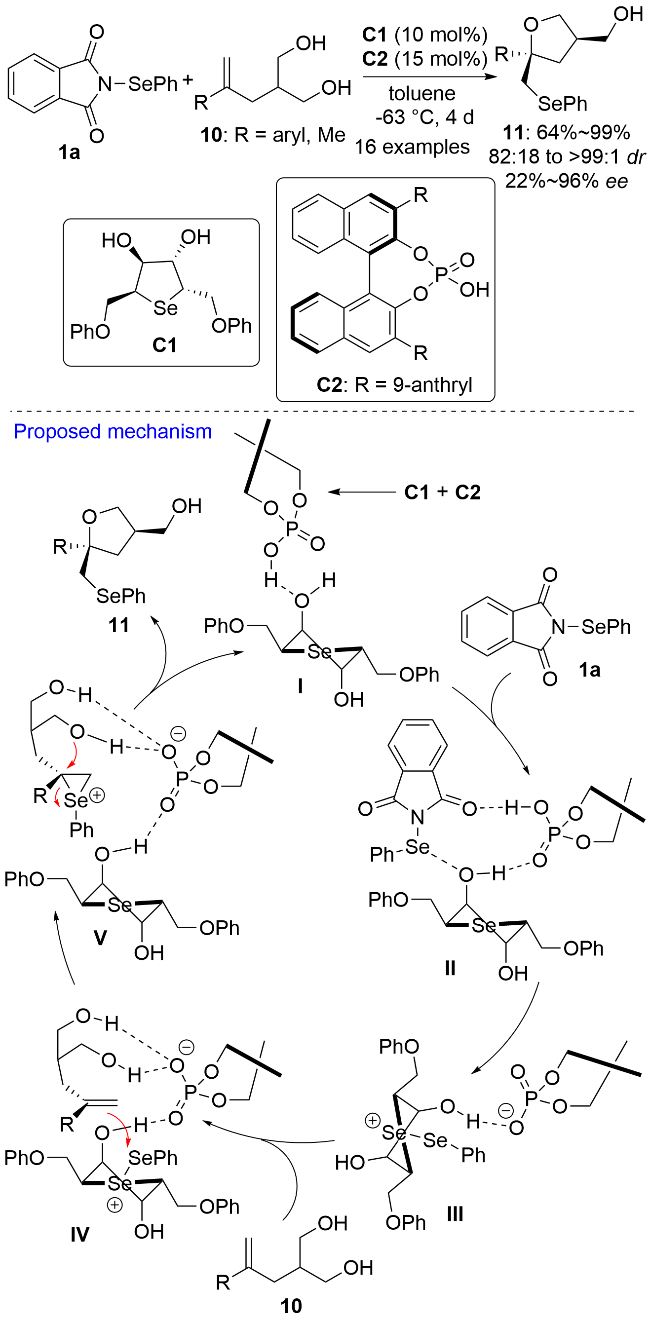
2010年, Denmark课题组[12]报道了首例手性路易斯碱催化的烯醇不对称硒化环醚化反应, 构建了手性β-芳硒基环醚分子, 但反应的对映选择性并不理想. 随后, 化学家们相继报道了多种手性有机小分子催化的烯烃不对称双官能团化反应(包括硒化酯化反应[13]、硒化氨基化反应[14]、硒化环醚化反应[15]和硒化内酯化反应[16]), 获得了一系列环状或开链结构的手性有机硒化合物. 2018年, Wong、柯志海和杨英洋等[17]以N-苯硒基邻苯二甲酰亚胺1a为亲电硒源, 利用手性路易斯碱C1和手性布朗斯特酸C2为共催化剂, 实现了不饱和1,3-二醇10的不对称去对称化硒化环醚化反应, 以最高99%的收率、>99∶1的dr值和96%的ee值获得目标手性有机硒化合物11 (Scheme 2). 值得注意的是, 当10上的R为邻位取代芳基时, 由于空间位阻过大而导致反应不能顺利进行. 作者结合密度泛函理论(DFT)计算提出了可能的反应机理: C1与C2通过氢键结合生成超分子I; I通过氢键和Se…O硫族键与1a结合生成中间体II; II 随即发生苯硒基正离子的转移生成中间体III; 烯醇底物10与III经中间体IV生成三元环硒鎓离子V; 之后在羟基的进攻下V发生开环反应生成产物11并重新生成超分子催化剂I进入下一个循环.
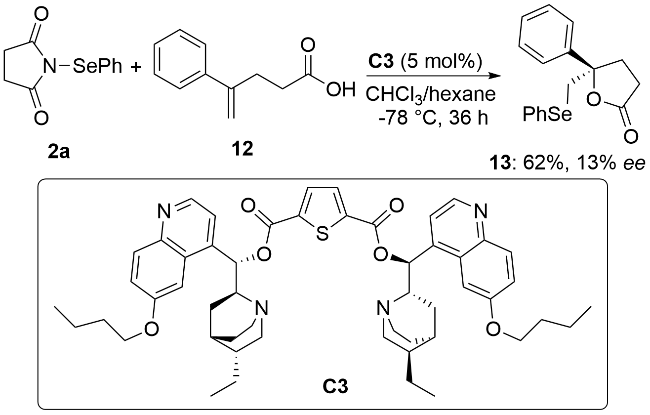
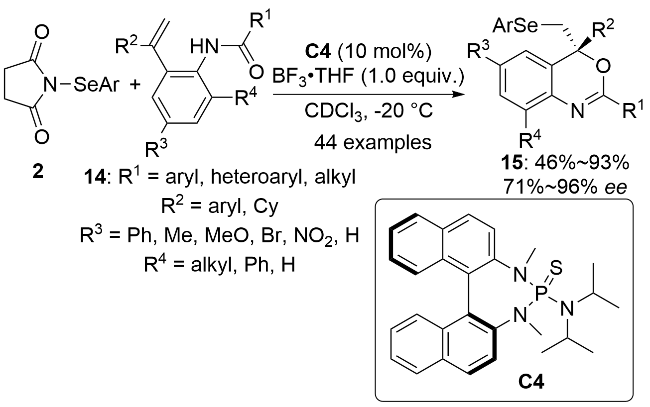
2018年, 闫海龙课题组[20]首次报道了在手性双功能方酰胺催化剂C5作用下, 硒代磺酸酯5与α,β-不饱和酮16的不对称硒化磺酰化反应(Scheme 5a). 该方法能够以良好的收率和对映选择性构建手性α-苯硒基酮17. 2019年, 秦文灵和毛辉等[21]也报道了一例手性催化剂C5催化的α,β-不饱和酮18的不对称硒化磺酰化反应(Scheme 5b). 该反应以饱和氯化钠水溶液为溶剂, 能够在温和条件下实现含有连续两个手性中心的有机硒化合物19的高对映选择性合成. 机理研究表明, 路易斯碱催化剂C5一方面与5结合, 另一方面作为氢键供体与18结合而形成中间体I, 活性芳硒基亲电试剂与烯烃亲电加成生成硒鎓离子中间体II, 反应中生成的磺酰基负离子随后对硒鎓离子进行亲核开环, 最终生成目标产物19并释放出催化剂完成催化循环.
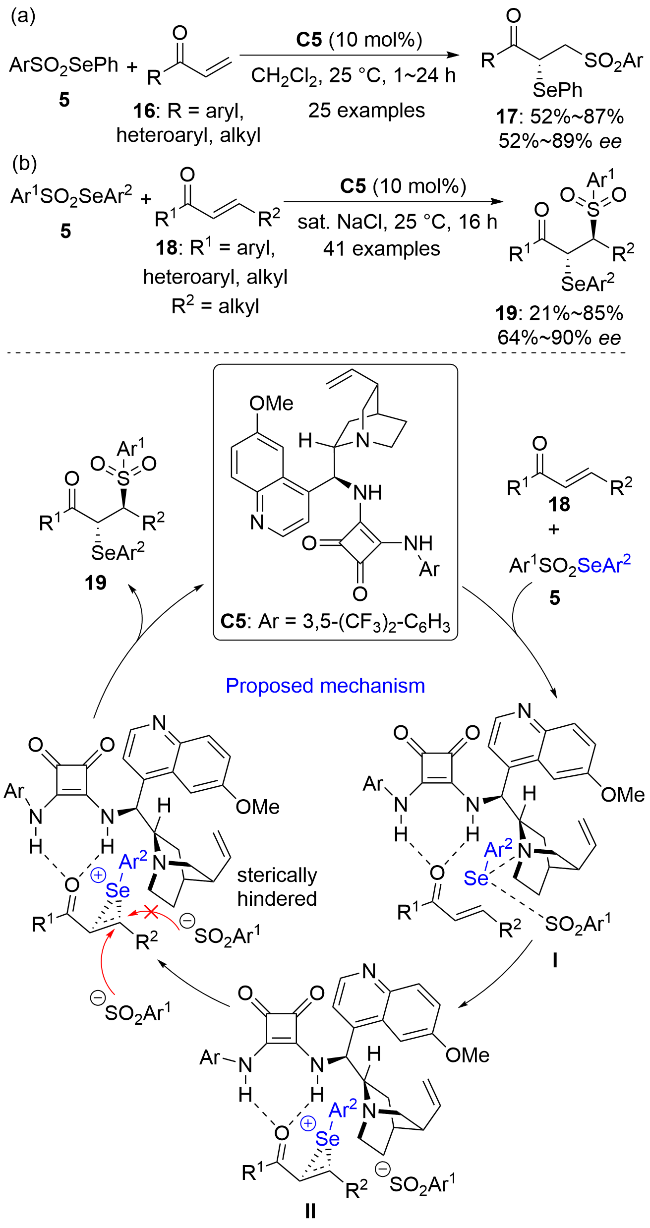
2025年, 柯华和陈志敏等[22]利用N-芳硒基糖精3作为亲电硒源, 首次实现了手性硫化物C6和非手性路易斯酸三氟化硼乙醚络合物(BF3•Et2O)共催化的1-(1-芳基乙烯基)环丁醇20的硒基化-半频哪醇重排反应, 以中等到优秀的收率和良好的对映选择性实现了含有手性季碳中心的α-芳硒基环戊酮21的合成(Scheme 6a). 值得注意的是, 使用邻硝基苯硒基亲电试剂是反应获得高对映选择性的关键. 苯环邻位上硝基的空间位阻、强吸电子效应以及硝基上氧原子与硒原子的硫族键弱相互作用能够有效降低亲核试剂对硒鎓离子中间体上硒原子的进攻, 从而抑制手性硒鎓离子中间体的外消旋化[12]. 将其换成苯硒基亲电试剂则可能会由于手性硒鎓离子中间体的快速外消旋化而导致反应的对映选择性急剧下降(如21a).
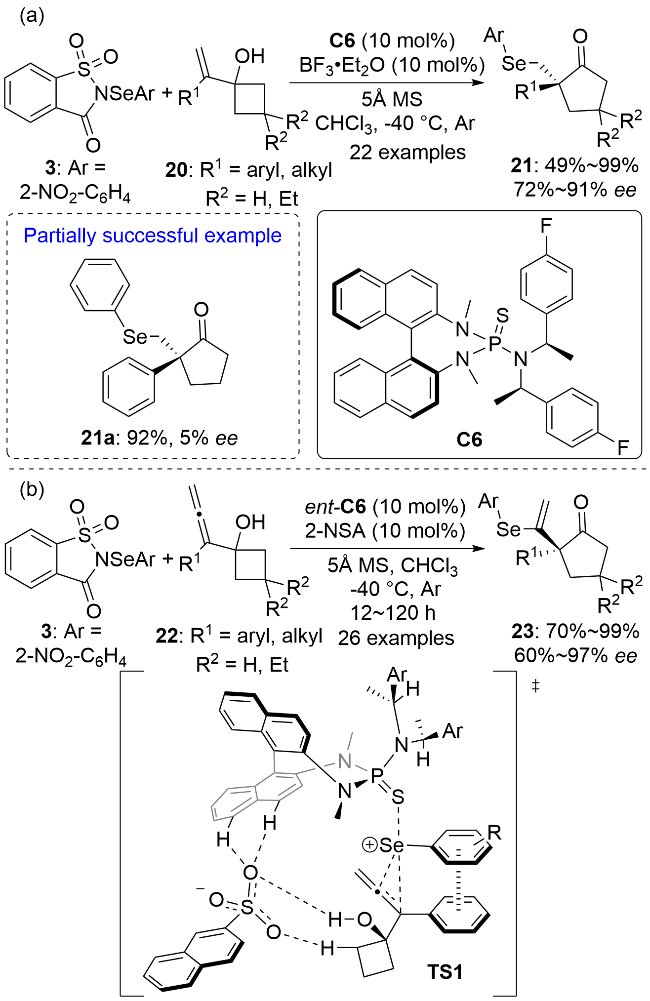
同年, 薛小松和陈志敏等[23]还报道了一例手性硫化物C6和非手性萘-2-磺酸(2-NSA)共催化的联烯基醇22的硒基化-半频哪醇重排反应(Scheme 6b). 共催化剂通过硫族键和氢键作用对亲电硒化试剂3进行活化, 以良好的区域选择性、化学选择性和对映选择性实现了手性α-(芳硒基乙烯基)环戊酮的合成. 作者通过过渡态TS1解释了反应的对映选择性. 活性亲电硒基化试剂从联烯醇22的Si面与联烯发生化学选择性亲电加成, 从而决定了产物的立体构型. 值得注意的是, 生成的三元环硒鎓离子中间体两个芳环间存在π-π堆积作用, 同时磺酸根负离子与催化剂C6以及22的环丁醇结构单元之间存在四个氢键. 随后, 1,2-迁移和亲核开环反应发生而生成最终的目标产物.
2.2 芳烃的阻转选择性硒基化反应
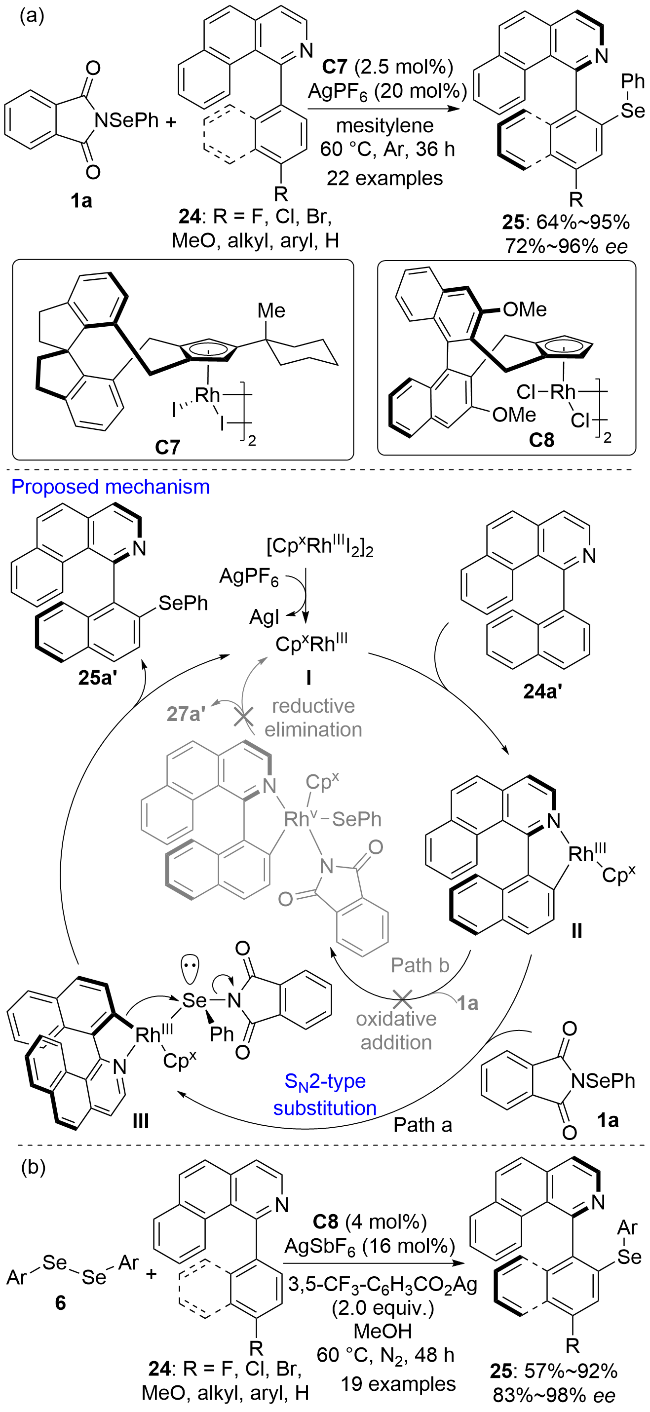
2024年, 郑超、顾庆和游书力等[24]使用三价铑络合物C7作为催化剂实现了1-芳基苯并[h]异喹啉24与N-苯硒基邻苯二甲酰亚胺1a的阻转选择性C—H键硒基化反应, 以高收率和高对映选择性得到系列轴手性的联芳基类有机硒化合物25 (Scheme 7a). 可惜的是, 当导向基苯并异喹啉变成异喹啉时, 反应的对映选择性很低, 这可能是由于异喹啉基团的空间位阻较小, 对反应过程中的手性诱导不利. 结合文献报道[25]、对照实验和DFT计算, 作者以1-(萘-1-基)苯并[h]异喹啉24a'的苯硒基化反应为例提出了可能的反应机理. 金属铑络合物对24a'进行配位活化后, 邻位C(sp2)—H键断裂生成手性五元环铑中间体II. 值得指出的是, 动力学同位素效应(KIE)实验表明决速步涉及C(sp2)—H键的断裂. 铑物种II与亲电硒化试剂配位后生成中间体III, 其C—Rh键电子对进攻硒原子, 邻苯二甲酰亚胺负离子从相反的方向离去, 通过上述SN2亲核取代反应生成了手性硒基化产物25a'. C(sp2)—H键断裂反应是可逆的, SN2反应的速率比环铑中间体的质子化反应速率要快, 从而促进了反应向硒基化反应的方向进行. 2025年, 于松杰和李兴伟等[26]以二芳基二硒醚6为亲电硒源也实现了类似的联芳基化合物的阻转选择性硒基化反应(Scheme 7b).
2.3 烯醇化合物的对映选择性硒基化反应
2.3.1 羰基化合物的对映选择性硒基化反应
2015年前的研究主要围绕着手性有机小分子催化醛的α-硒基化反应, 采用的是烯胺催化模式[27]. 2015年, 袁伟成课题组[28]利用非共价催化策略, 实现了辛可尼丁催化的3-吡咯基吲哚酮的不对称α-硒基化反应. 含羰基的氮、氧杂环化合物广泛存在于天然产物中, 往往具有生物活性, 是合成药物分子的重要结构单元[29]. 随后, 多种含羰基的氮、氧杂环化合物的不对称α-硒基化反应相继被报道. 2017年, Mukherjee课题组[30]报道了金鸡纳碱衍生物(DQ)2PHAL C9催化的丁烯内酰胺26的不对称α-苯硒基化反应(Scheme 8a). 手性产物27经一步反应即可方便地转化成手性γ-羟基内酰胺28, 它可以看作是26的不对称插烯羟基化反应的产物. 2020年, 伍新燕和韩建伟等[31]报道了手性膦亚胺C10催化的3-芳基吲哚酮29的不对称α-苯硒基化反应(Scheme 8b). 2022年, Waser课题组[32]报道了二氢奎宁C11催化的4-烷基-5(4H)-噁唑酮31和异噁唑烷酮33的不对称α-苯硒基化反应(Scheme 8c).
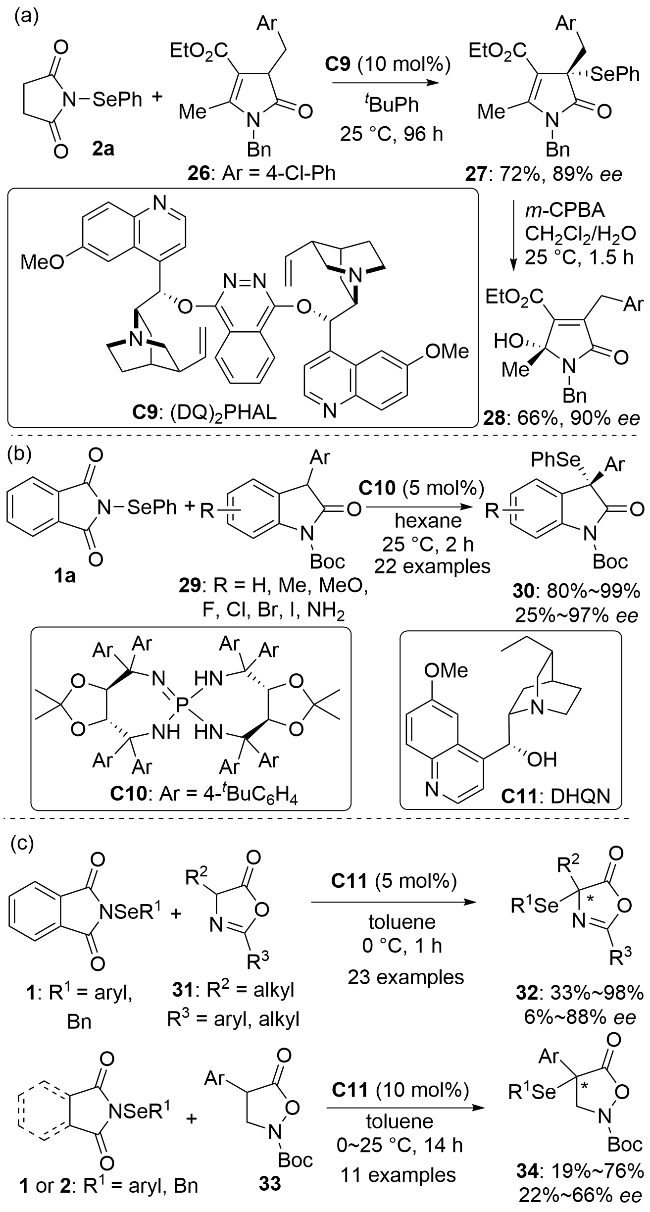
2019年, 陈甫雪课题组[33]使用N-硒氰基糖精4作为亲电硒源, 发展了Ni(OTf)2-(R,R)-DBFOX/Ph LI配合物催化的β-酮酸酯35的不对称α-硒氰化反应(Scheme 9). 作者提出了反应的立体控制模型, 底物35的酮羰基与镍配位活化后脱质子生成的镍-烯醇中间体与催化剂的苯环之间存在π-π堆积作用, 酯基端的空间位阻使得硒氰基正离子更有利于进攻烯醇中间体的Si面从而生成S构型的产物. 大位阻的金刚烷基(Ad)是反应取得高对映选择性的关键所在. 酯基部分的空间位阻减小(如甲基、异丁基)会导致反应的对映选择性下降. 该反应仅适用于六元环状β-酮酸酯, 当五元环状β-酮酸酯用作底物时, 只得到外消旋的硒氰化产物(如36c).
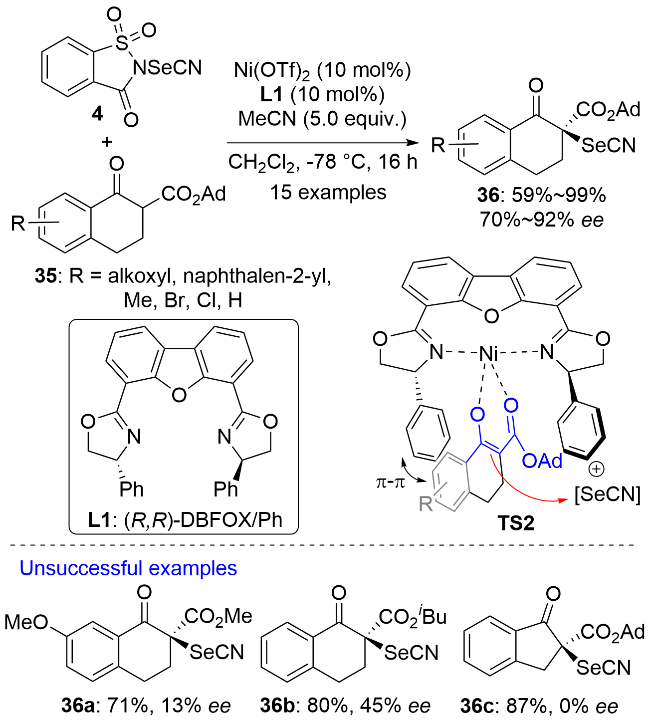
相对于环状羰基化合物而言, 开链结构羰基化合物的α-硒基化反应的立体选择性更难控制. 这是因为前者能够生成几何结构明确的烯醇式中间体, 而后者生成的烯醇式中间体的几何结构却很难控制. 2025年, 殷亮课题组[34]报道了Cu(I)-(S,S)-Ph-BOPA L2催化的2-酰基咪唑37的不对称α-硒基化反应(Scheme 10). 系列亲电试剂, 包括具有不同电性取代基的硒酚、杂芳硒酚及硒醇形成的硒代磺酸酯5都能被很好地兼容. 同时, 该反应表现出良好的官能团耐受性, 亲电试剂和亲核底物上的卤素、甲氧基、羟基和氰基等取代基对反应没有明显的影响. 遗憾的是, 该策略仅适用于α-单烷基取代的酰基咪唑, 当R1为芳基时, 生成的手性产物容易发生外消旋化而导致其光学纯度很差. 当酰基咪唑37羰基的α位连有两个取代基时, 反应不能发生.
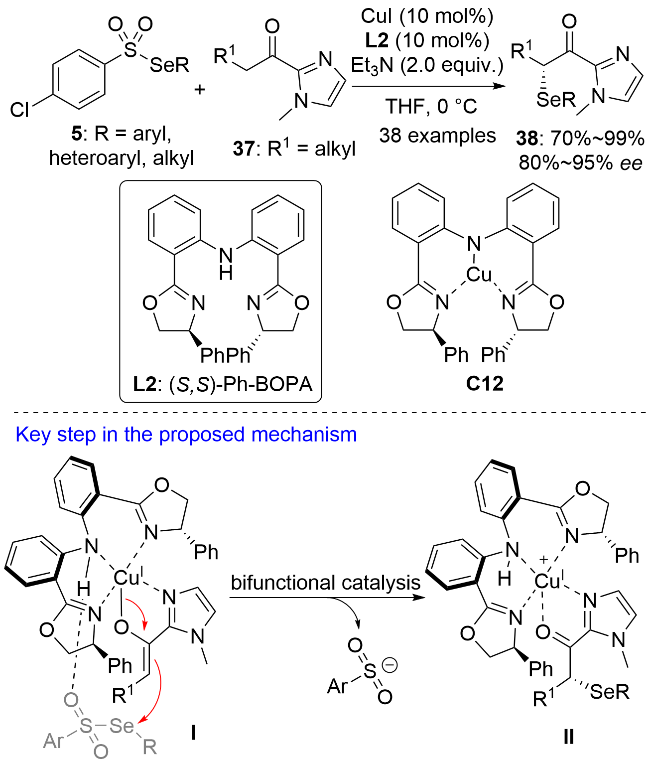
作者对反应机理进行了研究. 向反应体系中加入2,2,6,6-四甲基哌啶氧化物(TEMPO)、二丁基羟基甲苯(BHT)和1,1-二苯基乙烯时, 反应没有受到明显影响, 这说明该反应不是自由基机理; MesCu(I)作为铜源代替CuI/Et3N体系时, 获得了相同的实验结果, 这说明一价铜-烯醇物种是反应中的活性亲核试剂; 其它类型的亲电硒源(如苯硒基氯、二苯基二硒醚和N-苯硒基丁二酰亚胺)会导致产物的收率和ee值下降; 手性配体L2氮上的氢原子用甲基取代后也会导致反应的收率和ee值急剧下降. X射线光电子能谱(XPS)分析表明钳形一价铜配合物C12是反应中的活性催化剂. C12通过配位作用和氢键作用分别与烯醇中间体和硒代磺酸酯结合生成中间体I, 硒原子进攻烯醇中间体的Re面从而实现产物38的立体化学控制. 显然, 配体L2氮上的氢原子用甲基取代后将不能与硒代磺酸酯通过氢键作用相结合, 从而无法实现产物的对映选择性控制. 值得指出的是, 酰基咪唑是酯的合成等效物. 产物38可进一步转化为含有α-硒基的手性羧酸、羧酸衍生物和酮等.
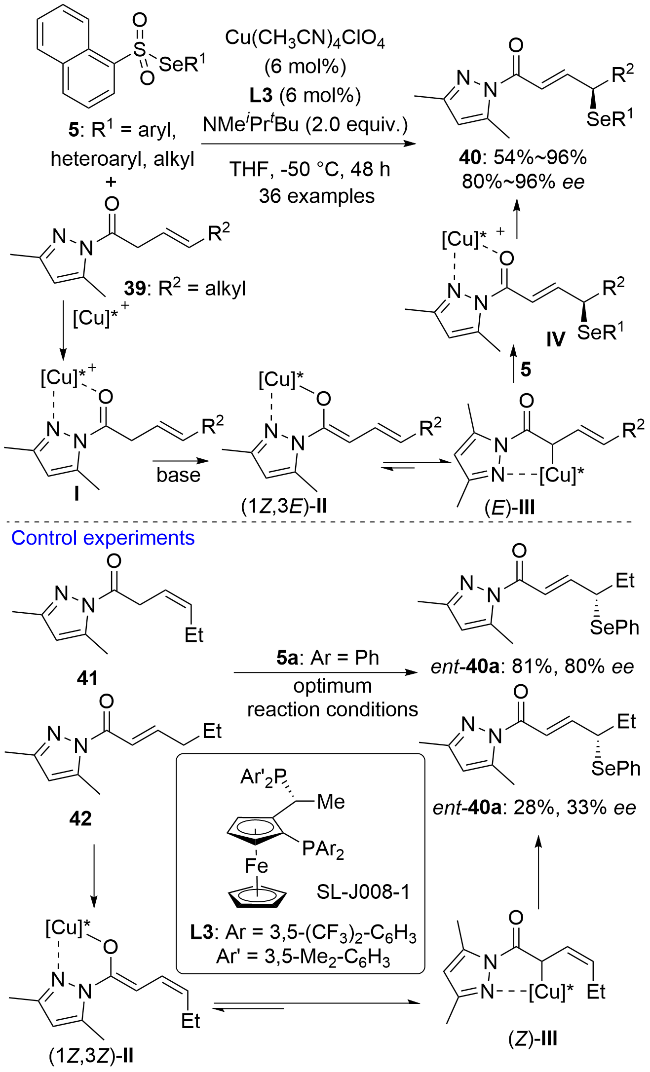
同年, 殷亮课题组[35]又报道了一价铜催化的(E)-β,γ-不饱和吡唑酰胺39与硒代萘-1-磺酸酯5的不对称插烯硒基化反应, 以高对映选择性构建了一类手性γ-硒基-(E)-α,β-不饱和吡唑酰胺骨架40 (Scheme 11). 该反应同样具有广谱的底物普适性和良好的官能团耐受性. 其它类型的亲电硒化试剂(如苯硒基氯和二苯二硒醚)可能由于具有较强的氧化性, 容易把一价铜催化剂氧化失活而导致反应收率降低甚至没有目标产物生成. 二烯醇中间体有α位和γ位两个亲核反应位点, 一般来说, 开链型二烯醇中间体更有利于在α位进行反应. 39与5在甲基异丙基叔丁基胺催化下的背景反应证明了这一结论, 而此反应则取得了优秀的γ选择性. 氘代动力学同位素实验结果表明, 一价铜的配位降低了39上α-H的pKa值, 这使得脱质子更容易发生. 同时, 手性配体SL-J008-1 L3展现出很强的配体加速效应. 在最优条件下, L3不仅有优秀的不对称诱导能力, 而且对反应的区域选择性控制起到了至关重要的作用. 亲核试剂前体39上咪唑2位的氮原子和羰基氧原子与一价铜配位活化后脱质子生成了二烯醇中间体(1Z,3E)-II和一价铜-烯丙基物种(E)-III, 后者在热力学上更稳定, 作为反应的活性亲核试剂与亲电硒源结合生成γ-硒基化产物. 反应过程中往往伴随着α-硒基化副产物的生成, R2基团的空间位阻越大, α-硒基化副产物的含量越高. 当R2为叔丁基时, 没有γ-硒基化产物生成. (Z)-β,γ-不饱和吡唑酰胺41作为亲核试剂前体脱质子生成的是(1Z,3Z)-II 和 (Z)-III, 后者与亲电硒源结合生成的是立体构型相反的插烯产物ent-40a. (E)-α,β-不饱和吡唑酰胺42作为亲核试剂前体生成的也是构型相反的产物, 但由于其γ-H的酸性较弱, 导致插烯硒基化反应活性较差, 产物ent-40a的收率和ee值均较低. 手性产物40经SN2或SN2′反应可以方便地转化为多官能团化的手性合成砌块.
2.3.2 亚烷基β-酮酸酯的不对称氧杂Michael加成-硒氰化反应
2.3.3 Se—S键与α-重氮酯的不对称卡宾插入反应
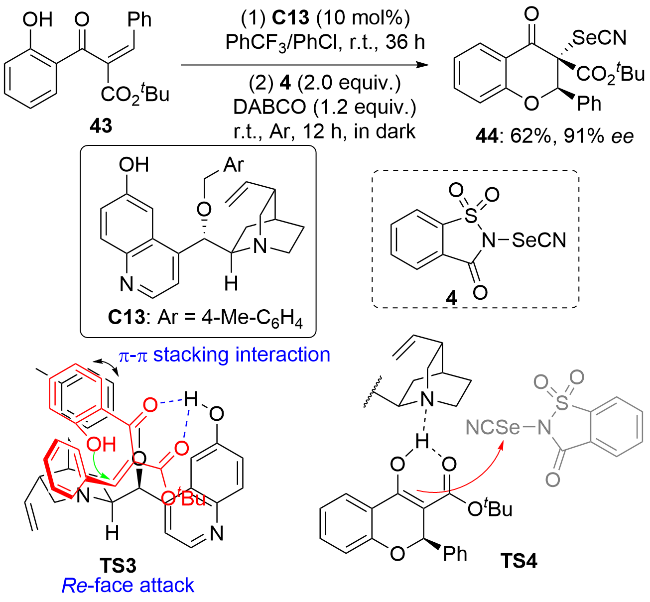
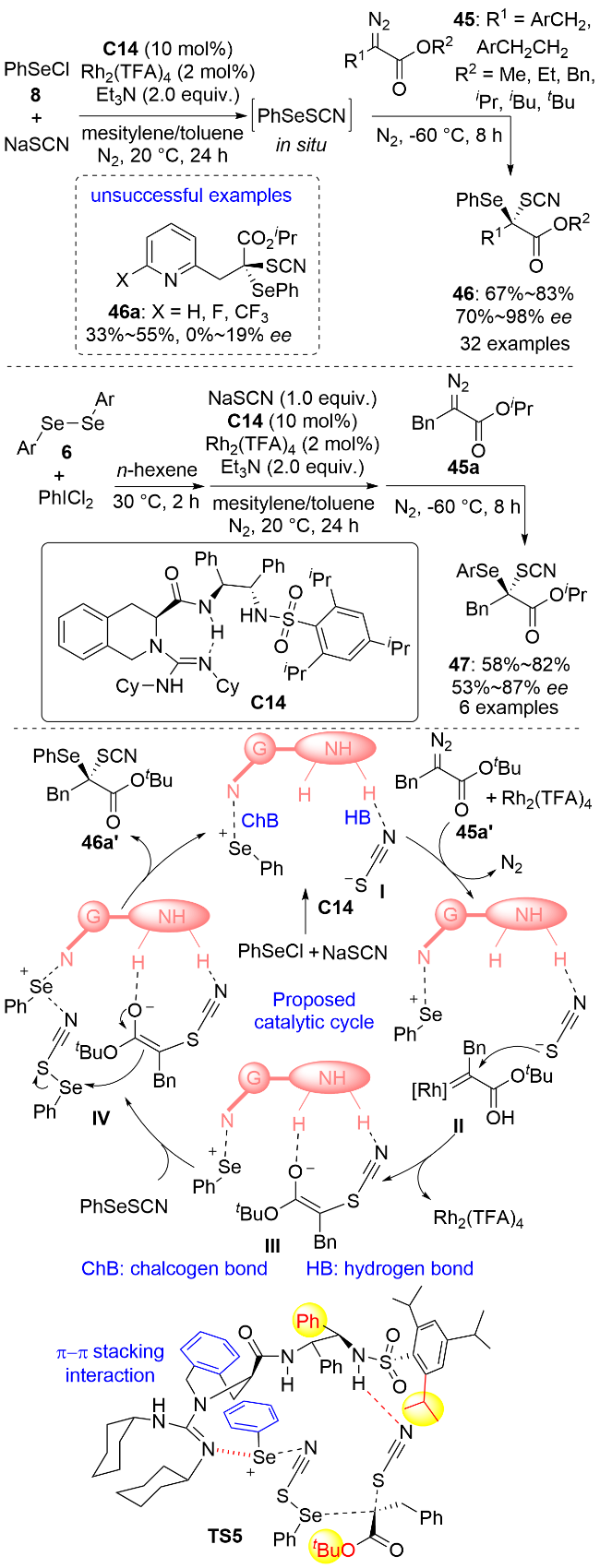
卡宾同时具有亲电和亲核双重反应活性, 因此可以同时在卡宾上引入两个杂原子. 2025年, 冯小明和刘小华等[37]利用苯硒基氯8和硫氰酸钠反应原位生成的PhSeSCN为硒源, 实现了二价铑/手性胍C14协同催化的α-重氮酯45的不对称卡宾插入反应, 以高对映选择性构建了同时连有苯硒基和硫氰基的碳手性中心(Scheme 13). 作者在条件优化过程中发现, 使用均三甲苯和甲苯混合溶剂以及1.5 equiv.的类卡宾底物45有利于反应获得较好的结果. 底物普适性研究表明, 一系列α-苄基(包括α-杂环芳基甲基)取代的α-重氮酯均能使反应得到较好的结果. 苯环上取代基的电性和位置对反应结果没有显著影响. 但是, 吡啶基由于氮原子可以和硒源形成硫族键, 这干扰了手性胍与硒源的结合从而导致反应的收率和对映选择性很低. α-芳基-α-重氮酯、α-甲基-α-重氮酯以及α位没有取代基的α-重氮酯均不适用于该反应. 由于芳硒基氯不稳定, 作者通过二芳基二硒醚6与二氯碘苯反应原位生成芳硒基氯, 从而通过四组分一锅三步法实现了系列手性有机硒化合物47的合成. 该反应有亲电加成、卡宾的插入及自由基反应三种可能的机理. 结合光谱实验和计算结果, 作者提出了可能的卡宾插入历程: 手性胍催化剂与PhSeSCN之间的氢键和硫族键相互作用使得PhSeSCN发生异裂形成中间体I, 然后硫氰酸根的硫端进攻高活性的金属卡宾物种II生成烯醇中间体III. 最后, 另一分子PhSeSCN通过硫族键活化后作为硒源与烯醇结合生成产物, 同时释放出中间体I而完成催化循环. 作者进一步用立体模型解释了反应的对映选择性. 底物酯基端的叔丁基、催化剂芳磺酰基苯环上的邻位异丙基及催化剂碳骨架上苯基空间上的排斥使得反应更有利于通过过渡态TS5进行, 从而生成R构型产物. 这类含硫手性有机硒化合物具有一定的生物活性, 在药物化学领域具有潜在的应用价值. 作者对手性产物进行了生物活性测试, 发现其具有较低的细胞毒性, 且对白介素-1β有明显的抑制作用.
3 亲核有机硒试剂与亲电底物的立体选择性反应
与亲核有机硒试剂反应的亲电试剂包括烯烃和炔烃、亚胺、α,β-不饱和羰基化合物、类卡宾和小环醚等(Scheme 14). 本节根据亲电试剂的不同, 分别展开综述.
3.1 烯烃和炔烃的立体选择性氢硒化反应
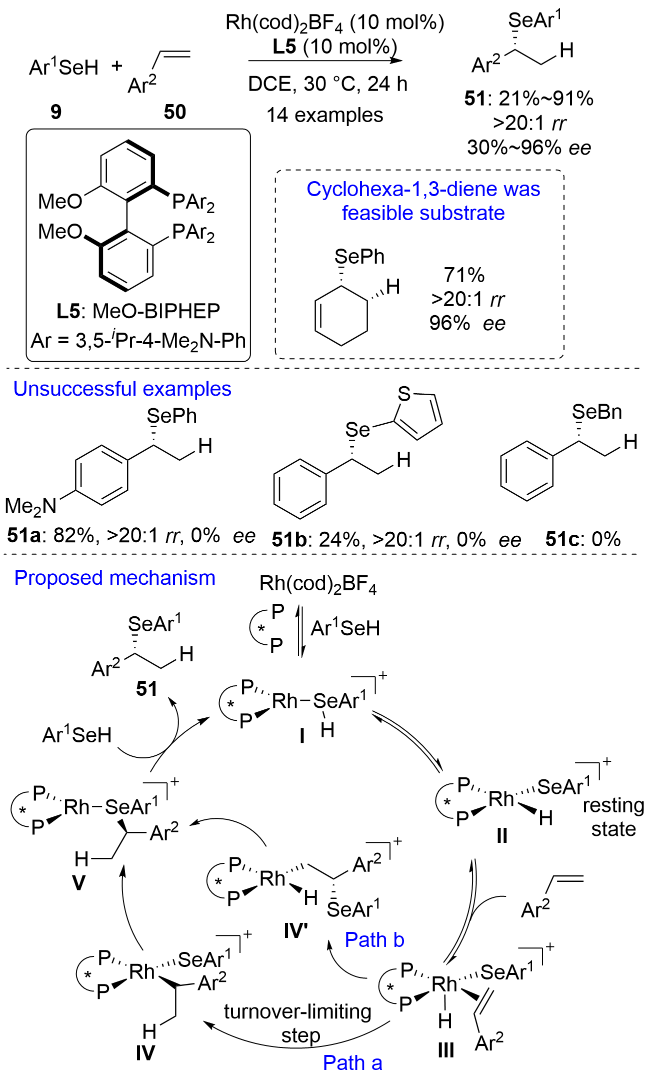
2022年, Dong和杨小会等[39]首次实现了一价铑催化的苯乙烯类化合物50的不对称氢硒化, 获得了符合马氏规则的支链型手性氢硒化产物51 (Scheme 16). 作者在最优反应条件下考察了苯环上含有不同电性取代基烯烃底物的适用性, 结果显示该反应对含有吸电子基和供电子基(二甲氨基除外)的苯乙烯均具有良好的兼容性. 邻位取代苯乙烯能获得优秀的对映选择性, 但由于空间位阻的影响产率较低. 环己二烯也适用于该催化体系. 但遗憾的是, 硒源的适用性不够宽泛. 该催化体系仅适用于硒酚, 不能兼容杂芳基硒酚和硒醇. 1H NMR研究发现, 硒酚对金属中心进行氧化加成后生成的铑-氢物种II是整个催化循环的静息态. 作者认为烯烃优先对Rh—H键进行迁移插入(Path a), 再发生C—Se键还原消除得到氢硒化产物. 迁移插入是整个催化循环的决速步, KIE值为1.8证实了这一结论. 随后, Furche、Dong和杨小会等[40]报道了光诱导烯烃的氢硒化反应, 通过自由基机理实现了反马氏加成, 但并未实现产物的立体选择性控制. 单春晖和龙榕等[41]利用DFT计算研究了铑催化苯乙烯不对称氢硒化反应的机理. 作者认为烯烃优先对Rh—Se键(Path b)而不是对Rh—H键进行迁移插入, 还提出了三层手性接力模型对反应的立体选择性进行了探究.
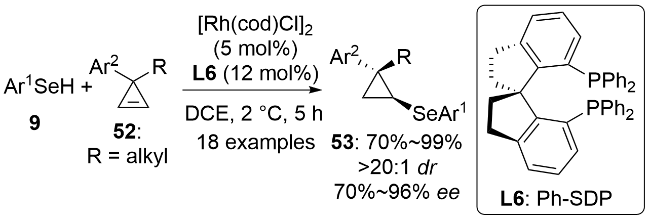
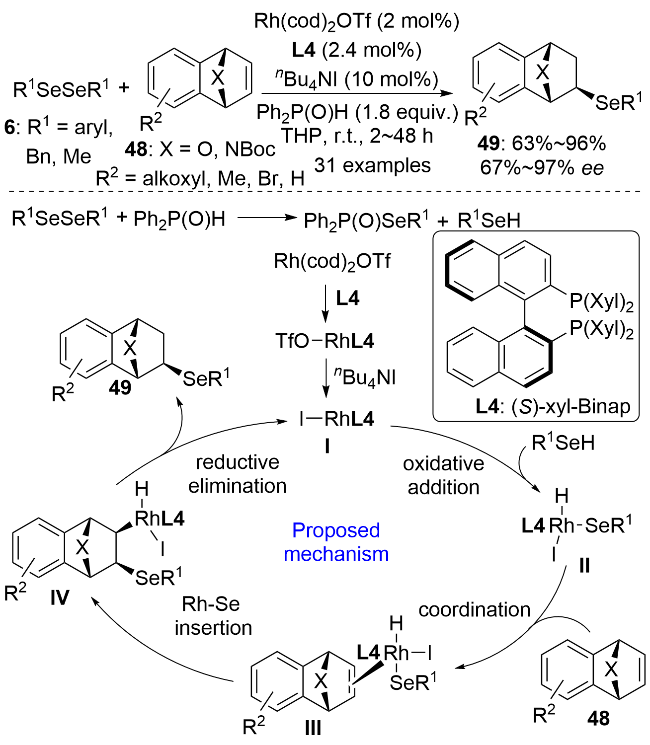
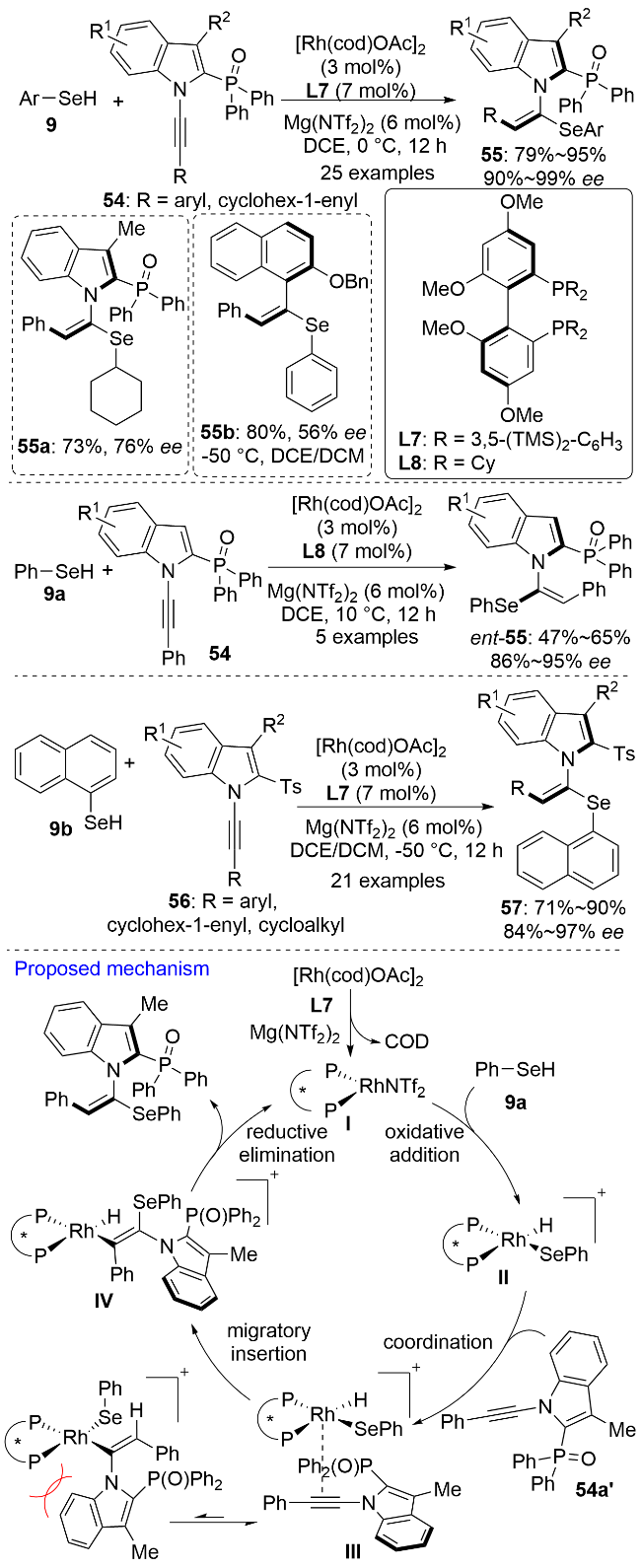
2024年, 李兴伟课题组[43]以硒酚9为硒源, 发展了一价铑催化的1-炔基-2-磷酰基吲哚54的阻转选择性氢硒化反应(Scheme 18). 该方法在温和的条件下, 以优秀的区域选择性、E式构型选择性和对映选择性合成了一系列的以碳氮键为手性轴的轴手性含硒化合物55. 这类轴手性的三取代烯烃是一类外消旋化能垒很低(约113 kJ•mol−1)的化合物. 特别需要指出的是, 二价镁盐Mg(NTf2)2的阴离子和阳离子对催化剂的活性都有至关重要的促进作用. 使用其它类型的镁盐或其他双三氟甲磺酰亚胺金属盐时, 反应效果均很不理想. 该催化体系不仅适用于硒酚, 还能兼容环己硒醇. 有意思的是, 使用手性配体L8和L7时得到的产物立体构型是相反的. 通过改变反应温度和溶剂等参数, 该催化体系还可以被拓展到1-炔基2-磺酰基吲哚56与萘-1-硒酚9b的氢硒化反应. 相较于磷酰基取代的底物而言, 磺酰基取代的底物反应活性更好, 但对映选择性稍低. 2-苄氧基-1-苯乙炔基萘也可以用作不对称氢硒化反应的底物, 但在其与苯硒酚的反应中只获得了56%的ee值. 结合机理实验和文献报道, 作者提出了可能的反应机理. [Rh(cod)OAc]2和手性配体L7在镁盐的协助下生成活性催化剂I. 反应中未观察到非线性效应, 这支持了铑与配体L7为1∶1的配位形式. I随后经氧化加成、迁移插入、还原消除生成最终的轴手性产物并再生催化剂完成催化循环. 在电子效应和空间位阻效应的共同作用下, 炔烃优先对Rh—Se键进行迁移插入生成中间体IV, 动力学实验表明这是反应的决速步. KIE实验(kH/kD=1.4)说明决定反应速率的关键步骤中不涉及Se—H键的断裂. R为缺电子芳基时, 炔烃底物的反应活性没有降低, 这说明Rh—H键的还原消除也不是反应的决速步.
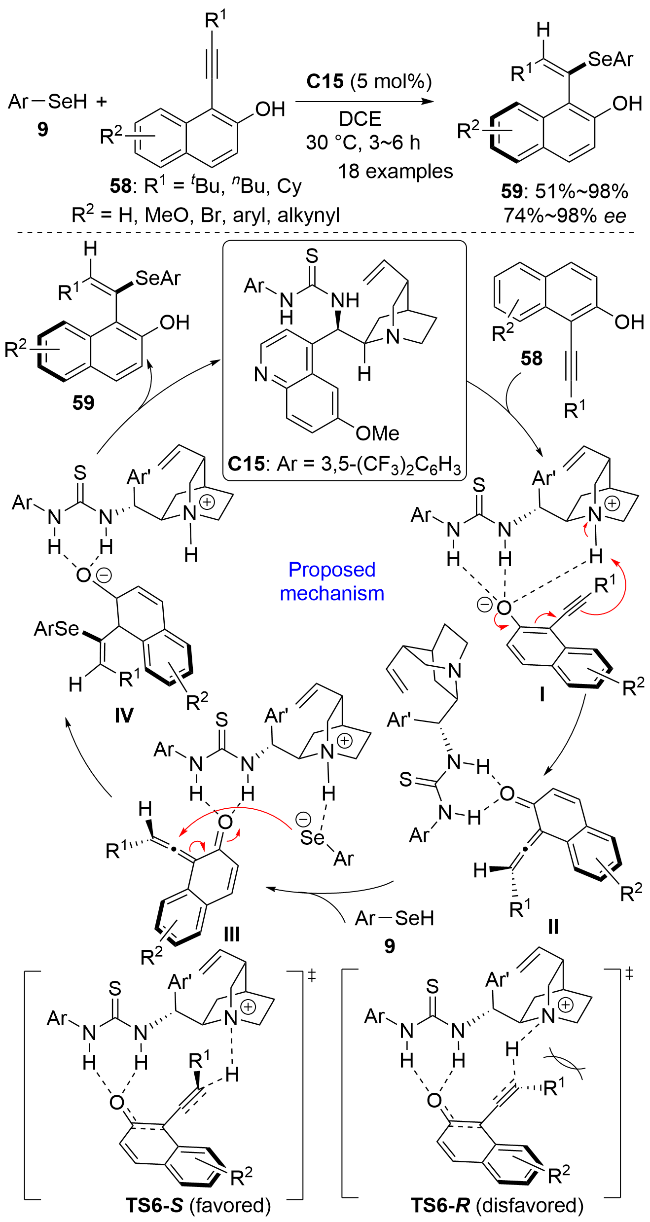
2025年, 卢宇和杨小会等[44]使用手性有机小分子催化剂C15实现了1-炔基萘-2-酚58的阻转选择性氢硒化反应, 同样在温和条件下实现了E式构型的轴手性乙烯基硒化合物59的高区域选择性和对映选择性合成(Scheme 19). 然而, 该催化体系仅适用于硒酚作为亲核硒源的情况, 当硒醇(如苄硒醇、环己硒醇)用作亲核试剂时反应则不能发生. 如图所示, 作者结合DFT计算提出了可能的反应机理. 在催化剂C15作用下, 炔基萘酚脱质子并发生质子转移生成S构型联烯醌中间体II. 硒酚与中间体II通过氢键结合并脱质子生成中间体III, 芳硒基负离子随后从联烯的E面(R1基团相反的方向)发生亲核加成反应生成中间体IV. IV经质子化后生成最终的E式S构型产物. 在过渡态TS6-S中, 萘酚底物与催化剂之间存在一系列C—H…π相互作用; 而TS6-R中萘酚底物与催化剂之间则存在空间上的互斥. 因而, 质子转移更有利于经由TS6-S进行, 从而实现对映选择性控制.
3.2 亲核有机硒试剂与酮亚胺的立体选择性亲核加成反应
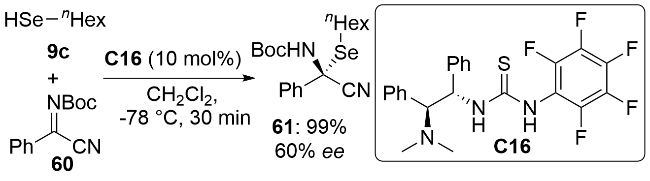
3.3 亲核有机硒试剂与α,β-不饱和羰基化合物的立体选择性Michael加成反应
硒醇或硒酚对α,β-不饱和羰基化合物的不对称Michael加成无疑是制备手性有机硒化合物最便捷的策略之一. 1979年, Wynberg等[47]报道了辛可尼丁催化的硒酚与环己-2-烯酮的不对称Michael加成反应, 这是首例亲核硒化试剂的不对称Michael加成反应, 但反应的ee值最高只有43%. 一方面, 由于硒醇(酚)的酸性较强, 容易脱质子生成亲核性较强的硒负离子, 从而导致背景反应的发生. 另一方面, 硒基负离子也是很好的离去基团, 这使得β-硒基取代的羰基化合物容易发生β-消除反应而回到原料. 因而, 硒醇(酚)与环己-2-烯酮的不对称Michael加成反应极具挑战性. 此后40多年再没有关于过渡金属或有机小分子催化的不对称共轭硒基化反应的报道.
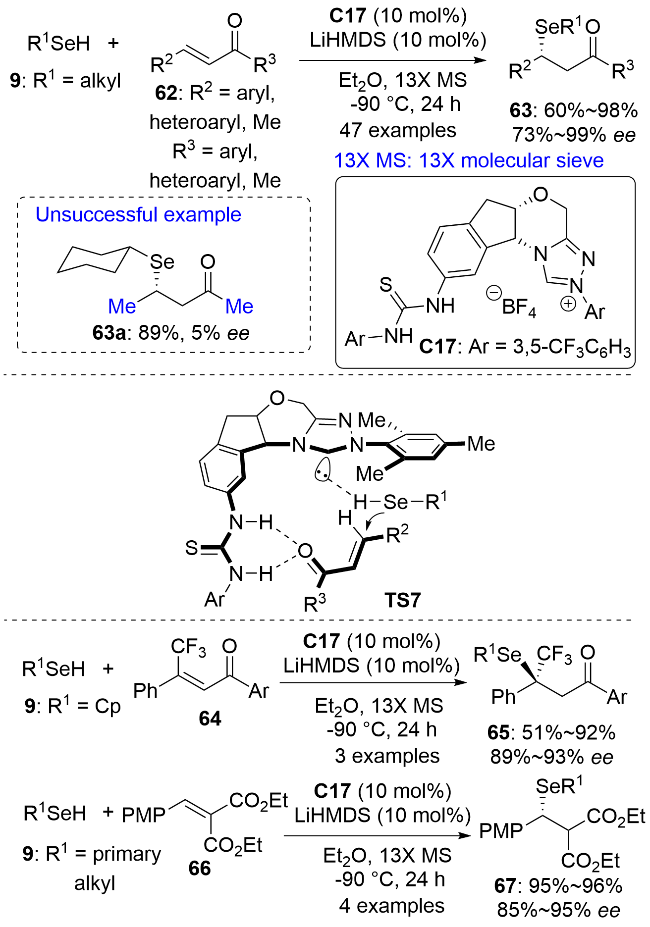
2022年, 陈杰安和黄湧等[48]报道了使用手性双功能氮杂环卡宾C17催化的硒醇9与α,β-不饱和酮62的对映选择性Michael加成反应, 以较高的对映选择性获得了系列手性β-硒基酮63 (Scheme 21). 机理研究表明, α,β-不饱和酮与催化剂的硫脲部分通过氢键结合, 与卡宾键合的硒醇从Re面进攻, 经过渡态TS7实现对产物手性的控制. 对硒源进行普适性考察发现, 仲烷基硒醇的对映选择性最好, 伯烷基硒醇可能由于较快的背景反应而导致反应对映选择性相对较低. 对Michael受体而言, R2和R3为多种电性的芳基均能有效兼容, 将其中一个基团换成甲基也能获得较好的反应效果, 但全部换成甲基则几乎生成外消旋产物. 该反应具有良好的官能团耐受性, 可用于复杂分子的后期不对称硒化修饰. 将β-三氟甲基查尔酮64用作Michael受体可以方便地构建同时连有三氟甲基和硒基的手性四取代碳中心. 该催化体系还可以实现α,β-不饱和二酯66与伯烷基硒醇9的高对映选择性Michael加成反应.
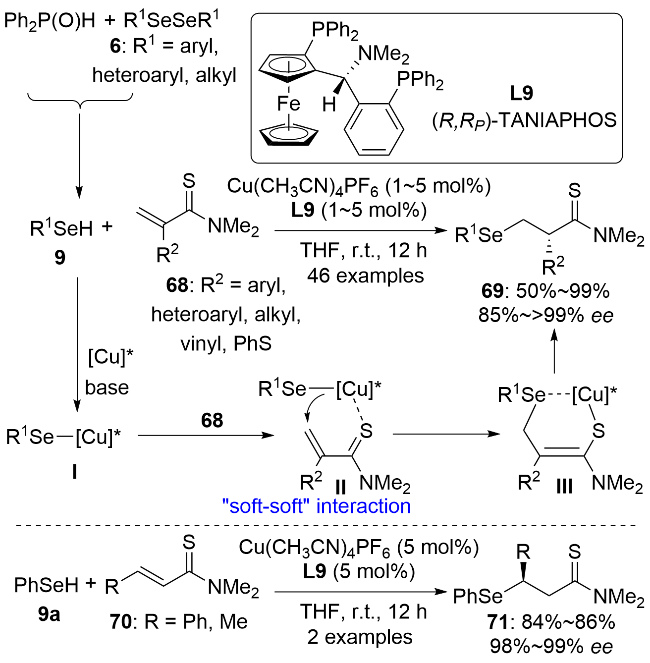
质子是体积最小的亲电试剂, 因此烯醇中间体的不对称质子化是一个具有挑战性的课题. 2023年, 殷亮课题组[49]使用一价铜-(R,RP)-TANIAPHOS L9催化体系催化α取代的α,β-不饱和硫代酰胺68与硒酚或硒醇9发生共轭加成反应, 通过烯醇中间体的不对称质子化反应合成了系列α手性的β-硒基硫代酰胺69 (Scheme 22).硫代酰胺68通过“软亲软”相互作用与一价铜配位, 同时亲核硒源9与一价铜形成铜-硒物种, 经六元环过渡态发生共轭加成反应, 生成的铜-硫代烯醇物种通过硒与铜的配位形成六元环中间体III, 这对硫代烯醇一价铜中间体起到了稳定作用, 从而使得接下来的对映选择性质子化反应能高选择性地发生. 控制实验表明, 等物质的量的Cu(CH3CN)4PF6与苯硒酚混合后会部分转化成铜-苯硒物种, 再加入等物质的量的手性双膦配体L9后会全部转化成铜-苯硒络合物物种, 这说明手性配体的配位使得铜盐的碱性显著增强. 因此, 反应体系中不需要额外加入弱碱(如三乙胺). 该反应具有广谱的底物适用性, 能兼容多种R1和R2为芳基、杂环芳基和烷基的亲核底物和亲电底物. 0.5 mol%催化剂载量的克级规模制备反应很好地说明了该反应的实用性. 该催化体系还可以被拓展到β取代的α,β-不饱和硫代酰胺70与苯硒酚9a的共轭加成反应中, 以优秀的对映选择性生成了β手性β-硒基硫代酰胺71. 作者通过X射线衍射法测得{[Cu-(R)-TOL-BINAP]-SePh}2物种中Cu—Se键的键长为0.248 nm, 这证实了反应过程中有Cu—Se键的生成.
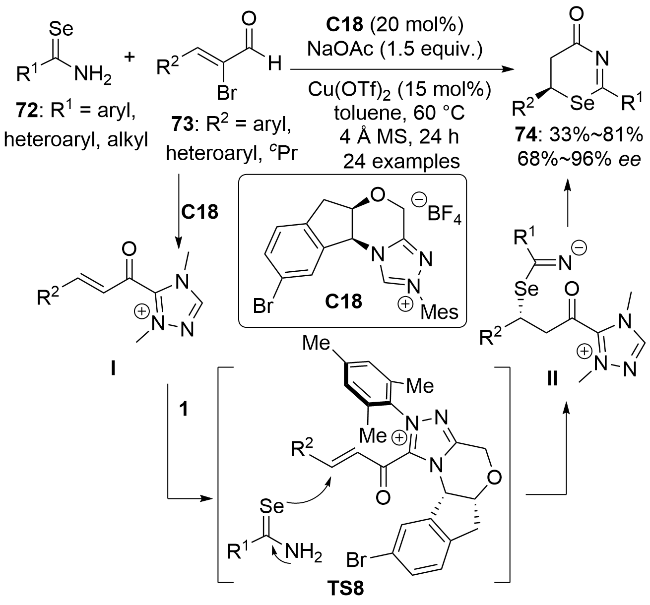
3.4 亲核有机硒试剂与类卡宾的立体选择性反应
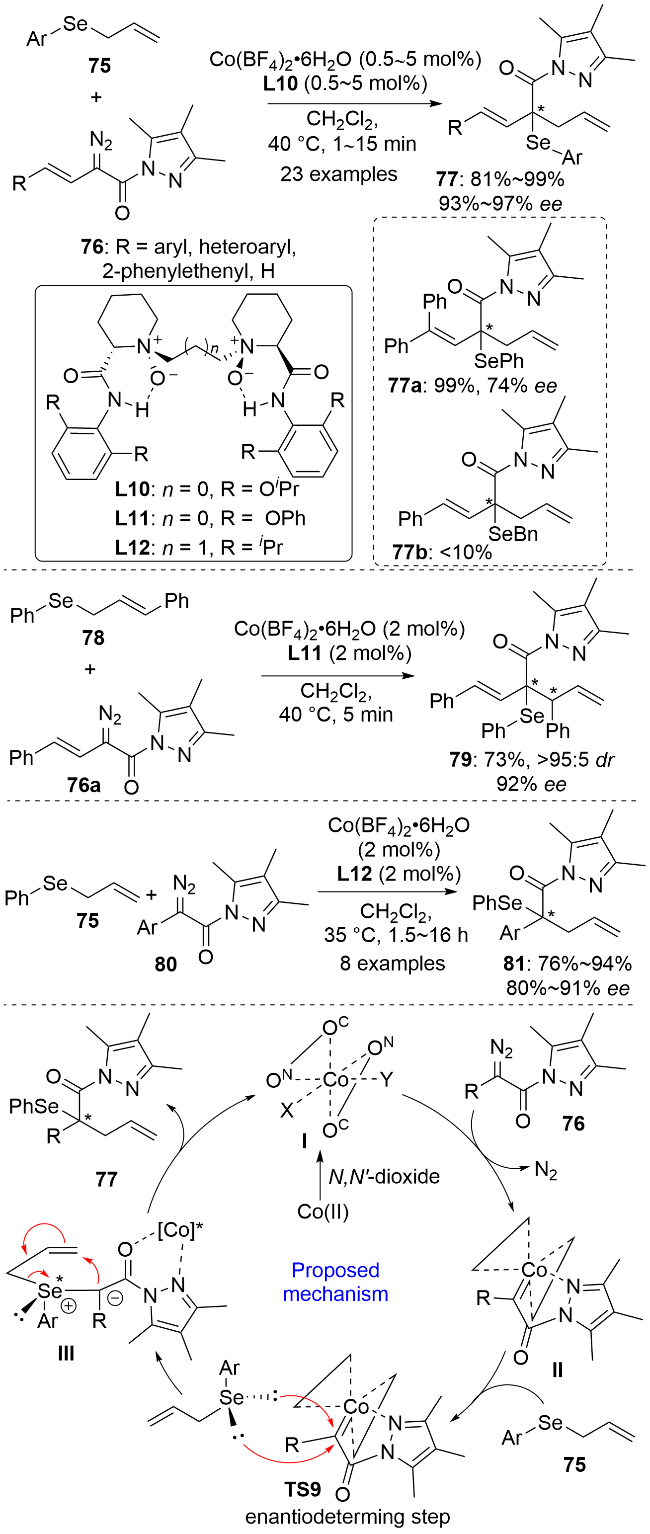
2020年, 刘小华和冯小明等[51]利用二价钴/冯氏手性双氮氧配体L10催化体系, 在温和条件下实现了烯丙基硒化合物75与乙烯基取代的α-重氮吡唑酰胺76的立体选择性[2,3]-σ迁移反应(Scheme 24). 作者在亲电底物普适性考察中发现, 当76上的R为连有吸电子基和供电子基的芳基、杂环芳基、苯烯基和氢原子时, 反应均能在很短的时间内顺利完成. 空间位阻较大的γ位二苯基取代的底物也可以几乎定量转化成相应的产物77a, 但反应的ee值明显下降. 在对亲核底物的普适性考察中发现, 该反应体系仅适用于烯丙基芳基硒醚, 不能兼容烯丙基苄基硒醚. 以烯丙基硒化合物78为亲电试剂可以实现含有连续两个手性中心的硒化合物79的高非对映选择性和对映选择性合成. 同时, 亲电试剂还可以拓展到芳基取代的α-重氮吡唑酰胺80, 反应同样具有广谱的底物普适性. 苯环对位或间位上含有吸电子基时产物的对映选择性更高, 但同时反应时间也更长. 遗憾的是, 当Ar为邻位取代芳基时可能由于空间位阻过大而导致反应不能顺利进行. 该反应的机理如下: 首先, 在二价钴配合物催化下重氮化合物脱去氮气生成卡宾物种II; 随后, 烯丙基硒化合物在手性配体的诱导下对卡宾物种进行立体选择性的亲核加成生成硒手性的叶立德中间体III; 最后, III发生[2,3]-σ迁移反应, 经五元环过渡态将手性中心从硒原子转移到碳原子上, 生成最终的手性硒产物.
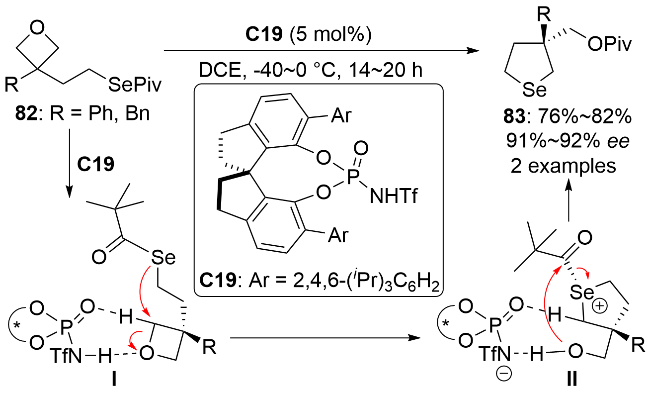
3.5 亲核有机硒试剂与小环醚的立体选择性开环反应
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4 总结与展望
含硒试剂与相应底物的催化不对称C—Se成键反应是合成手性有机硒化合物最便捷和最经济的策略之一. 我们对近十年来(2016年至今)该领域的研究进展进行了简单的总结. 研究人员开发了一系列不同的过渡金属/手性配体催化剂和手性有机小分子催化剂, 成功实现了亲电或亲核硒试剂与多种不同底物的不对称C—Se成键反应, 构建了多种碳手性和轴手性的有机硒化合物. 这些工作为含硒手性有机小分子催化剂和具有药物活性的含硒手性分子的发展提供了有效的不对称催化合成途径, 促进了合成化学、药物化学等相关领域的发展, 展现出重要的应用价值.
虽然催化不对称构建手性有机硒化合物的方法在近年来获得了快速的发展, 但是该领域的研究仍然存在显著的局限性. 首先, 从反应机制的角度出发, 目前尚缺乏催化不对称自由基硒化反应的报道. 显然, 自由基反应的特性将为手性有机硒化合物的合成带来新的区域选择性和新的反应模式, 这对创制新的手性功能有机硒化合物至关重要. 其次, 从反应手段的角度出发, 目前的催化不对称硒化反应还停留在传统的热化学模式, 使用新的反应手段(如光催化和电催化)不仅将带来新的反应模式, 还将提高特定反应的效率和选择性. 再次, 从过渡金属催化剂的类型来说, 贵金属铑的催化在不对称构建手性有机硒化合物中占据重要地位. 因而, 一方面用廉价金属催化替代贵金属铑催化的不对称硒化反应, 一方面利用廉价金属的特性发展独特的催化不对称硒化反应, 它们将成为手性有机硒化学发展的一个主要方向. 最后, 目前的硒化试剂比较有限, 还存在稳定性较差等问题(如一些烷硒基试剂). 因而, 发展结构新颖且兼具反应活性和稳定性的有机硒试剂显得非常重要. 我们相信, 经过化学家们的不断努力, 随着手性催化剂和不对称催化策略的不断发展, 更多反应条件温和且具有广泛底物普适性的催化不对称构建手性有机硒化合物的新方法必将被发展出来, 进而推动不对称合成、药物化学和农药化学等领域的发展.
Cheng, B.