


1 引言
2 结果与讨论
2.1 催化剂的表征
2.1.1 催化剂的形貌
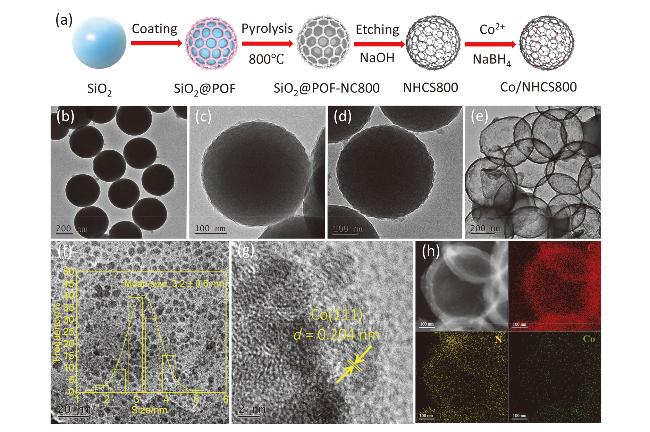
图1 (a) Co/NHCS800的合成示意图; (b~e) SiO2、SiO2@POF、SiO2@POF800、NHCS800的TEM图像; (f) Co/NHCS800的TEM图像和相应粒径分布图; (g) Co/NHCS800的高分辨TEM图像; (h) Co/NHCS800的高角度环形暗场扫描透射显微镜(HAADF-STEM)及能量分布面扫描分析(EDS mapping)图像Figure 1 (a) Schematic of Co/NHCS800 synthesis; (b~e) TEM images of SiO2、SiO2@POF、SiO2@POF800、NHCS800; (f) TEM image of Co/NHCS800 and the corresponding particle size distribution. (g) High-resolution TEM image of Co/NHCS800; (h) HAADF-STEM and energy distribution surface scanning analysis (EDS mapping) images of Co/NHCS800 |
2.1.2 催化剂的物相分析
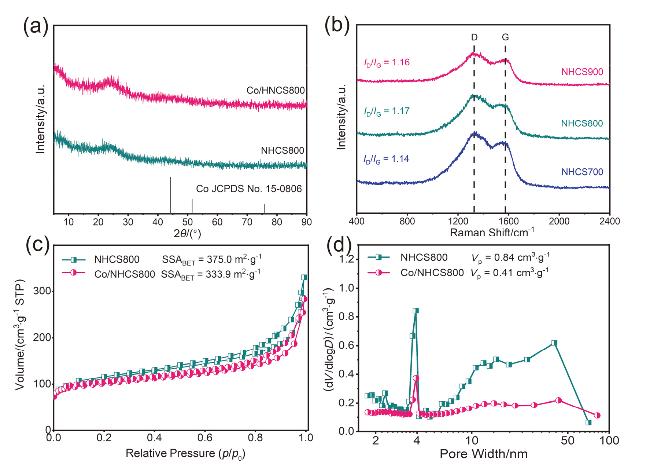
图2 (a) NHCS800和Co/NHCS800的XRD谱图; (b) NHCS700、NHCS800和NHCS900的Raman谱图; (c) NHCS800和Co/NHCS800的N2吸 附-脱附等温曲线; (d) NHCS800和Co/NHCS800的孔径分布图Figure 2 (a) XRD patterns of NHCS800 and Co/NHCS800; (b) Raman spectra of NHCS700, NHCS800 and NHCS900; (c) N2 adsorption-desorption isothermal curves of NHCS800 and Co/NHCS800; (d) Pore size distribution curves of NHCS800 and Co/NHCS800 |
2.1.3 X射线光电子能谱(XPS)分析
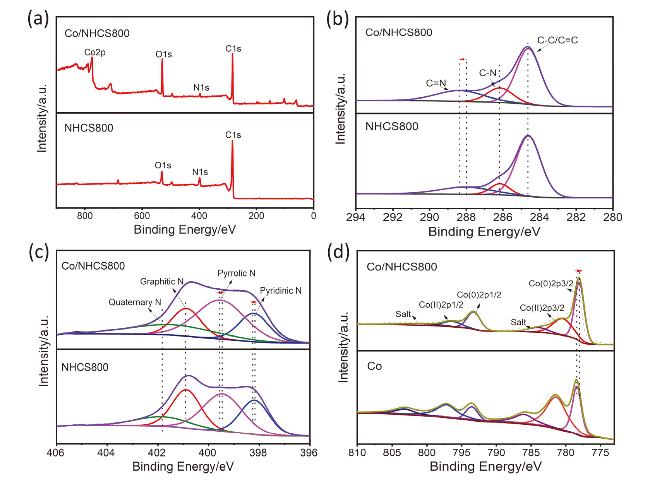
2.2 催化氨硼烷醇解性能研究
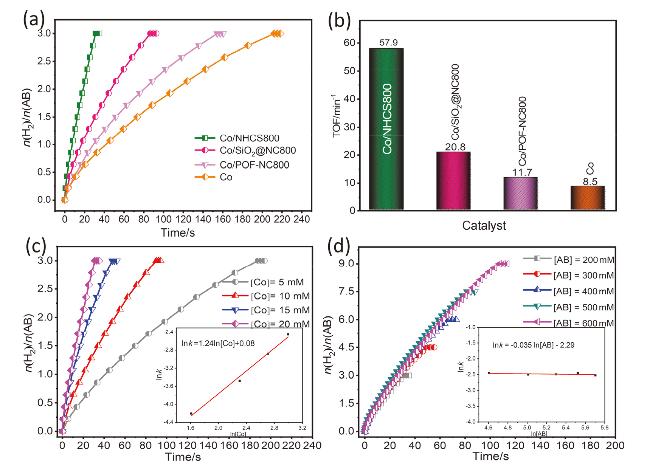
图4 (a) Co负载不同载体催化剂催化氨硼烷醇解制氢性能; (b)相应TOF值; (c)不同催化剂初始浓度下Co/NHCS800催化氨硼烷醇解制氢性能及ln [Co] vs. ln k拟合线; (d)不同氨硼烷初始浓度下Co/NHCS800催化氨硼烷醇解制氢性能及ln [AB] vs. ln k拟合线Figure 4 (a) Time course plots of H2 generation for the methanolysis with different catalyst; (b) The corresponding TOF value; (c) Time course plots of H2 generation for methanolysis of AB over Co/NHCS800 with different initial catalyst concentrations and ln [Co] vs. ln k plot.; (d) Time course plots of H2 generation for methanolysis of AB over Co/NHCS800 with different initial AB concentrations and ln [AB] vs. ln k plot |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
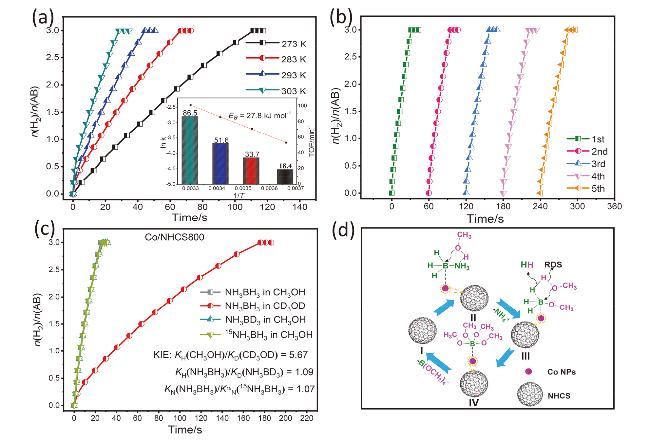
图5 (a)不同温度下Co/NHCS800催化氨硼烷醇解制氢性能和相应的Arrhenius曲线及TOF值; (b) Co/NHCS800催化氨硼烷醇解制氢的循环稳定性测试; (c) Co/NHCS800催化氨硼烷醇解的同位素标记实验; (d) Co/NHCS800催化AB醇解机制Figure 5 (a) Time course plots of H2 generation from the methanolysis of AB over Co/NHCS800 at different temperature and the corresponding Arrhenius plot and TOF value; (b) Durability test for the methanolysis of AB over Co/NHCS800; (c) Isotope-labeled experiments for methanolysis of AB catalyzed by Co/NHCS800; (d) Possible mechanism of AB methanolysis catalyzed by Co/NHCS800 |