


偶氮化合物是一类含有偶氮基(—N=N—)的有机化合物, 通过偶氮基可以连接两个相同或不同的芳香环, 也可以连接其他类型的基团. 这种结构赋予了偶氮化合物独特的性质, 使其在有机合成[1]、生物医药[2]以及食品工业[3]等领域都有着广泛的应用. 其中, 偶氮二羧酸酯由于酯基的存在可以参与多种类型的反应. 例如偶氮二羧酸酯具有亲电性, 可以被亲核试剂进攻发生迈克尔加成[4]. 其次, 偶氮二羧酸酯可作为氧化剂, 用于氧化醇、肼、硫醇和羟胺等化合物[5]. 除此之外, 由偶氮二羧酸酯和三苯基膦反应生成的Huisgen两性离子中间体[6]可与缺电子烯烃、醛酮及亚胺等化合物发生环加成, 为构建氮杂环提供了一种简单有效的方法. 相较于偶氮二羧酸酯, 偶氮苯类化合物因连有芳环而性质存在差异. 一方面, 芳环的存在扩展了偶氮苯类化合物的共轭体系, 使得其具有更强的吸光性. 偶氮苯类化合物能够通过光激发实现顺式和反式异构体之间的转换, 广泛用作光响应材料[7]. 另一方面, 偶氮基团的存在不仅能提高芳环的反应活性, 还能控制反应的位点选择性, 从而大大提高了偶氮苯类化合物在有机合成中的应用. 例如偶氮苯可在过渡金属催化下与各种偶联伙伴[8](烯烃、炔烃、醛、重氮酯和叠氮化物等)通过C(sp2)—H官能化构建氮杂环, 该策略目前已成为构建相应杂环的有效手段.
可见光作为一种清洁的可再生能源, 具有丰富易得和低碳环保的优点. 近年来, 人们对环境友好型合成方法的需求日渐增长, 因此可见光促进的有机合成反应受到了越来越多的关注[9]. 相较于传统的加热反应, 可见光促进的反应操作便捷、易于控制, 能在温和的条件下实现化学键的断裂与形成. 迄今为止, 可见光促进的反应已成为设计和开发多种反应的重要平台, 展现出优异的合成价值和应用潜力.
鉴于近期已有综述总结了芳基偶氮砜参与的光化学转化[10], 因此本文主要聚焦可见光促进偶氮二羧酸酯和偶氮苯类化合物参与的光化学转化, 并对该领域所面临的挑战以及未来的发展方向进行探讨.
1 N—H键的构建
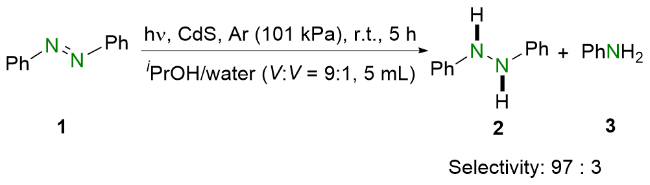
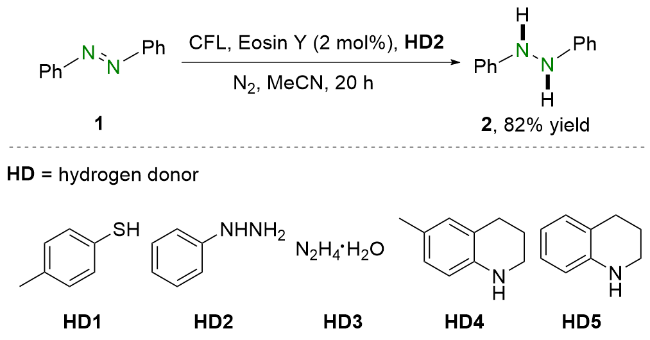
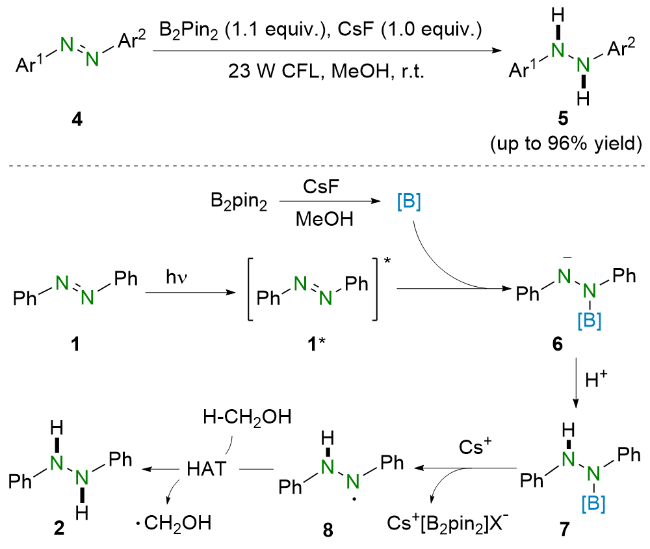
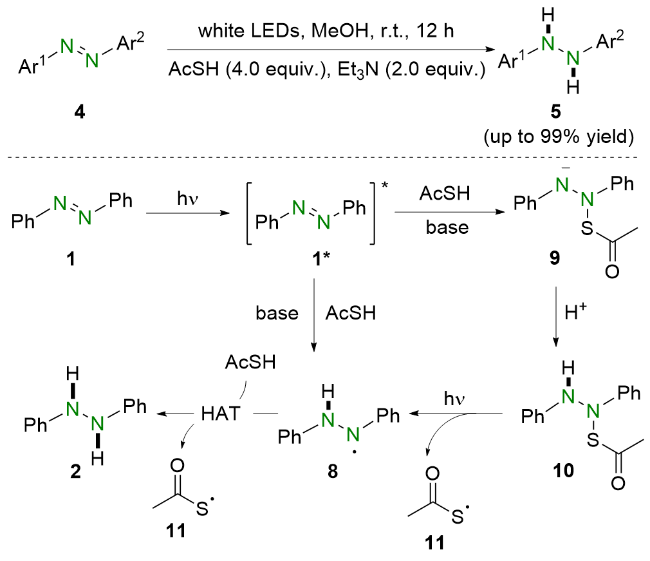
2021年, 王喜存和杨靖亚等[15]报道了将可见光与联硼酸频哪醇酯(B2Pin2)结合还原偶氮苯类化合物的策略(Scheme 3). 与之前的报道相比, 他们所提出的方法简便高效, 并且具有良好的官能团耐受性, 多种偶氮苯均能以良好至优异的收率转化为相应的氢化偶氮苯. 基于实验结果和相关文献报道, 作者提出了可能的反应机理. 在可见光照射下, 偶氮苯1到达激发态1*. 随后, 激发态的偶氮苯1*与原位生成的硼物种作用得到氮阴离子中间体6. 之后, 氮阴离子6在甲醇的作用下质子化生成硼氢化中间体7, 并进一步脱硼得到氮中心自由基8. 最后, 该自由基中间体8从甲醇中攫取氢原子生成氢化偶氮苯2.
2 C—N键的构建
2.1 氢酰化反应
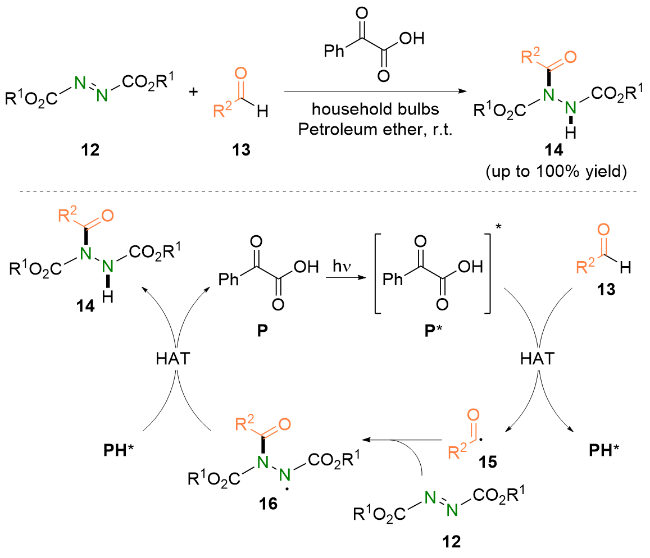
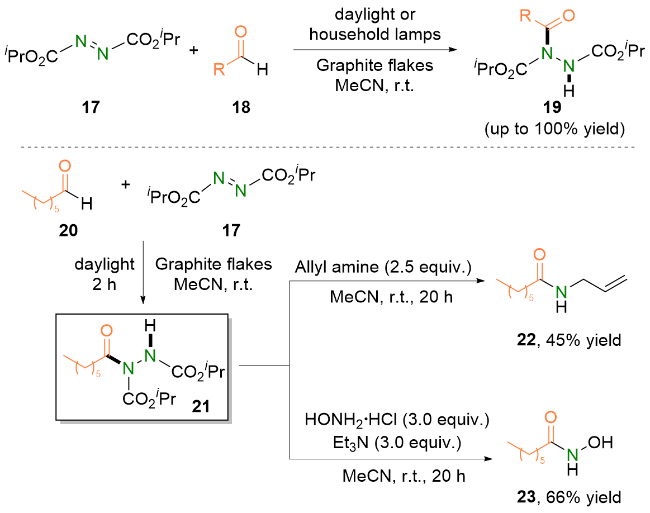
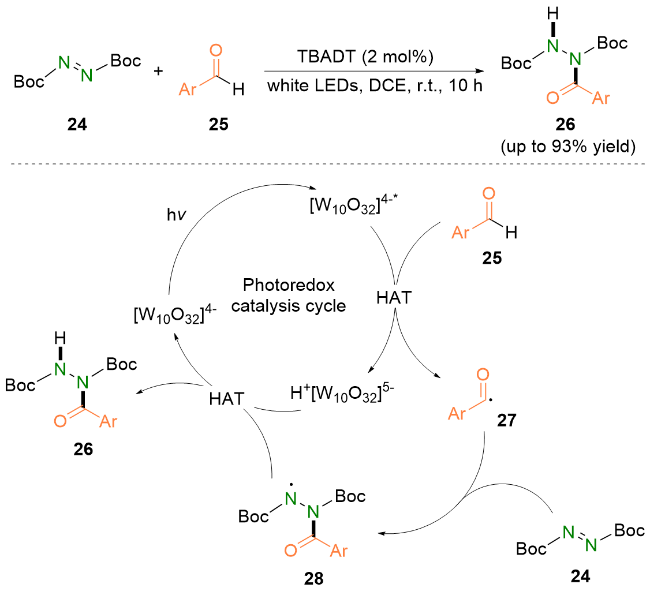
2023年, Kokotos等[22]报道了可见光加速的偶氮二羧酸酯与醛的氢酰化反应. 他们使用水作为溶剂, 仅在紫光照射下即可在极短的反应时间内(15~210 min), 以优异的收率将多种芳香族醛和脂肪族醛转化成对应的酰肼类化合物.
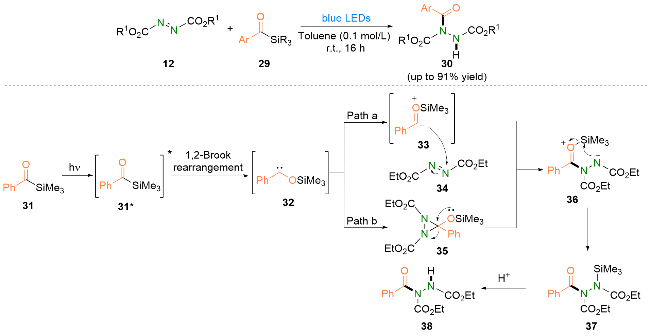
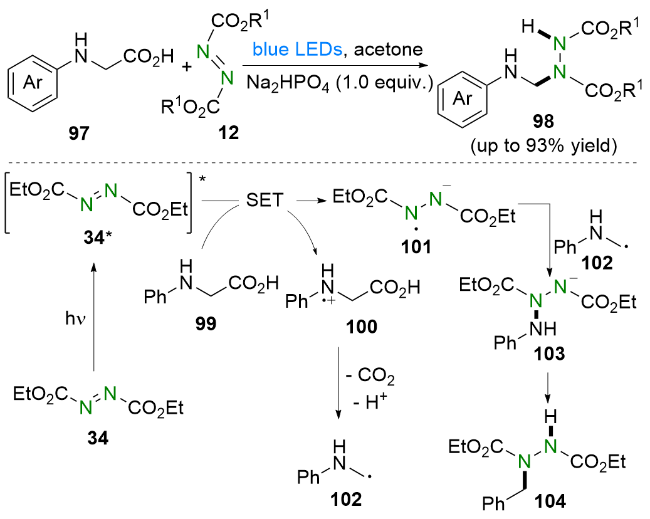
2024年, Yadagiri等[23]报道了偶氮二羧酸酯和酰基硅烷在可见光促进下合成酰肼类化合物的策略(Scheme 8). 基于实验结果和相关文献, 他们提出了可能的反应机理. 首先, 激发态的酰基硅烷31*发生1,2-Brook重排生成硅氧卡宾32. 第一种可能的途径: 硅氧卡宾32共振成为两性离子对33, 随后进攻偶氮二甲酸二乙酯(DEAD, 34)生成中间体36. 第二种可能的途径: 硅氧卡宾32与DEAD发生[2+1]环加成生成三元环中间体35, 不稳定的中间体35进而发生开环, 得到中间体36. 接着硅基从氧原子向氮原子迁移生成中间体37. 最后, 随着硅基的离去得到氢酰化产物38.

除了偶氮二羧酸酯以外, 偶氮苯类化合物同样可以参与酰肼类化合物的合成. 2020年, 霍聪德和杨靖亚等[25]报道了可见光介导的偶氮苯类化合物与α-酮酸的脱羧氢酰化反应(Scheme 10). 该反应成功进行的关键之处在于蓝光能够激发具有光活性的偶氮苯到达激发态. 此外, 该反应无需外加任何催化剂和添加剂, 并且CO2是该反应唯一的副产物. 根据实验结果和相关文献, 他们认为, 偶氮苯1首先在光照条件下到达激发态1*. 随后, 激发态的偶氮苯1*与苯甲酰甲酸P发生单电子转移, 致使苯甲酰甲酸P氧化脱羧生成酰基自由基42. 之后, 酰基自由基42会与自由基阴离子41偶联生成氮阴离子中间体43. 最后, 该阴离子中间体43再经过一步质子化即可得到偶氮苯的氢酰化产物44.
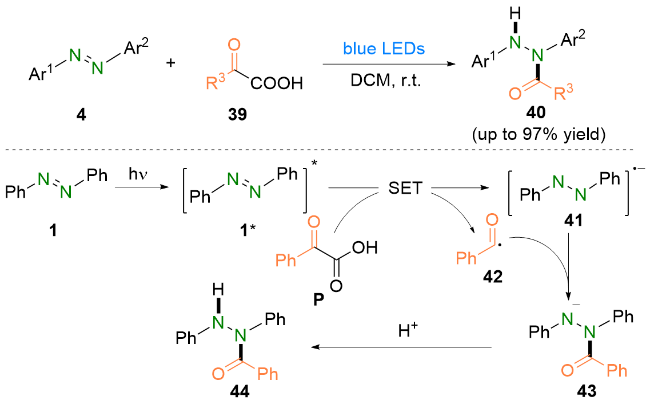
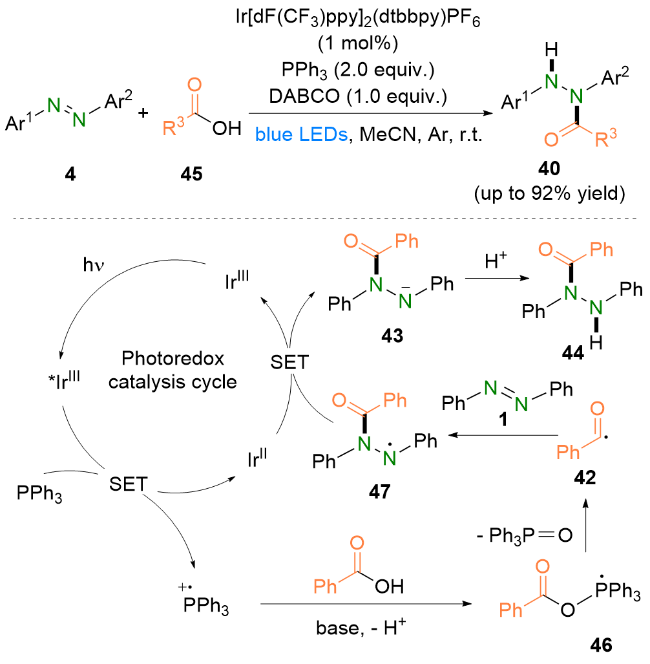
机理研究和相关文献表明, IrIII在光照下到达激发态IrIII*, 随后被三苯基膦还原猝灭得到IrII, 并生成三苯基膦自由基阳离子. 与此同时, 苯甲酸在碱的作用下脱质子生成苯甲酸根, 进而被三苯基膦自由基阳离子捕获得到磷中心自由基46, 再经历一步β-断裂生成酰基自由基42. 随后, 酰基自由基42与偶氮苯1发生自由基加成生成氮中心自由基47. 之后, 自由基中间体47氧化IrII得到氮阴离子43并再生IrIII. 最后, 阴离子中间体43经过质子化即可得到偶氮苯的氢酰化产物44. 几乎是同一时间, 夏远志等[27]也报道了类似的偶氮苯氢酰化策略.
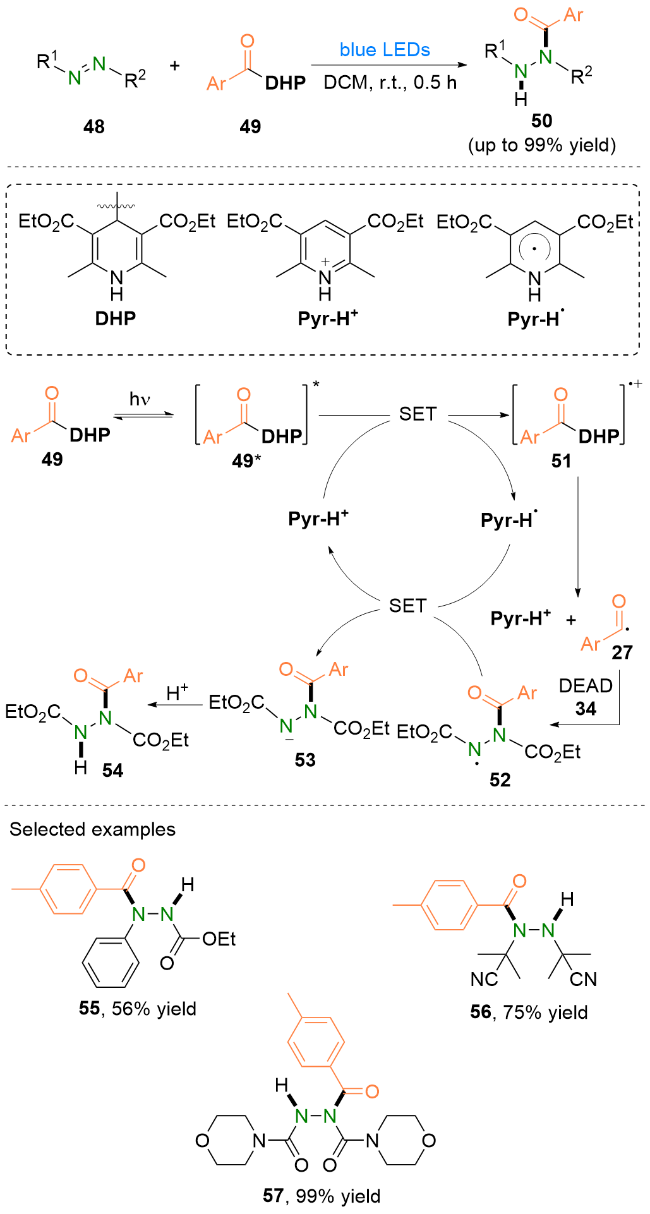
作者提出了反应机理为: 首先, 4-酰基取代汉斯酯49在蓝光的照射下到达激发态49*, 随后被单电子氧化成为自由基阳离子51; 之后, 不稳定的自由基阳离子51发生C—C键均裂生成Pyr-H+和酰基自由基27; 产生的酰基自由基中间体27则被DEAD 34捕获生成氮中心自由基52; 随后, 自由基中间体52与Pyr-H•发生单电子转移生成氮阴离子53, 再经历质子化即可得到氢酰化产物54.
2.2 氢烷基化反应
烷基自由基是最基本的活性中间体之一, 通常由脂肪族羧酸、硼试剂和卤化物等自由基前体产生, 目前已被广泛应用于有机合成[29].
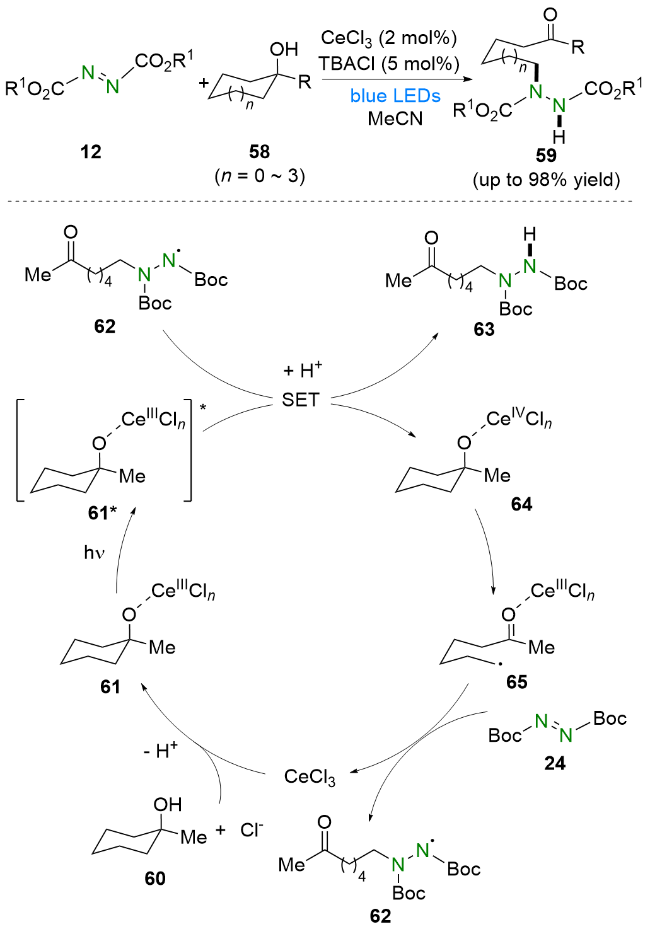
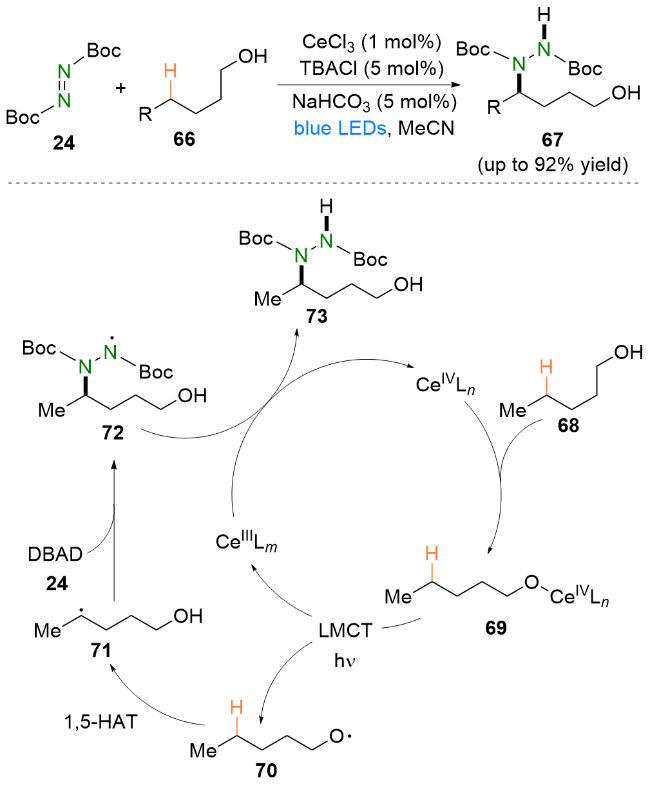
在烷基自由基中, α-氨烷基自由基因其活性高和易获得等优点一直以来受到化学工作者的广泛关注[34].
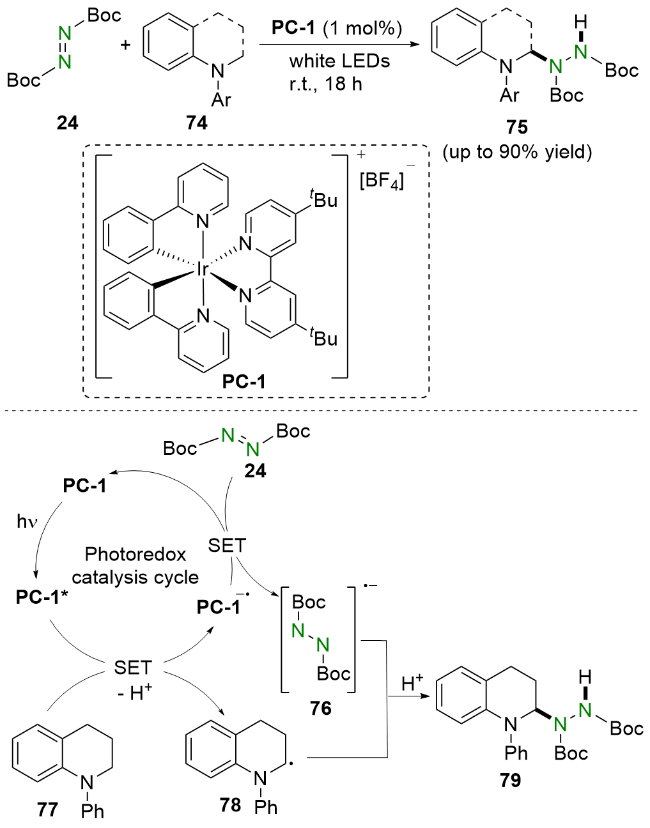
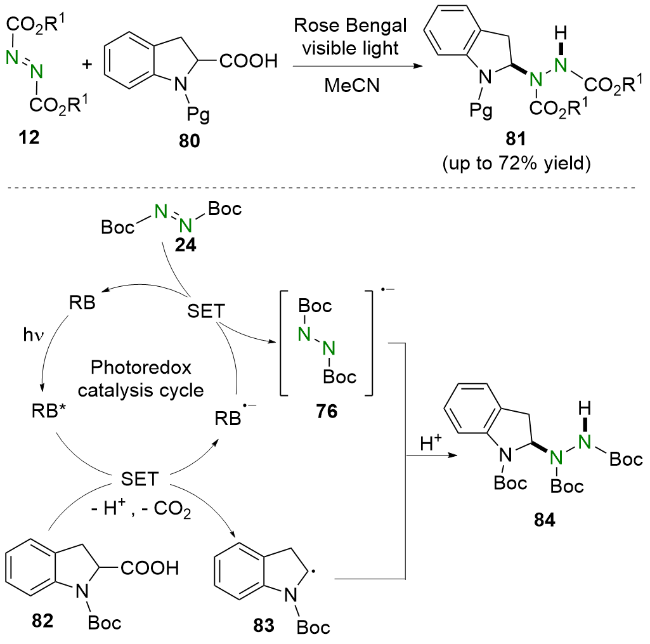
值得一提的是, 该反应还可以拓展到其他含α-氧原子和N-Boc-四氢异喹啉羧酸的环状体系. 机理研究和相关文献表明, RB在可见光照射下到达激发态RB*, 随后被N-Boc-吲哚-2-羧酸82还原猝灭并产生α-氨烷基自由基83. 同时, DBAD 24被RB-•还原生成自由基阴离子76. 最后, 自由基阴离子76与α-氨烷基自由基偶联83, 再经过质子化生成氢烷基化产物84.
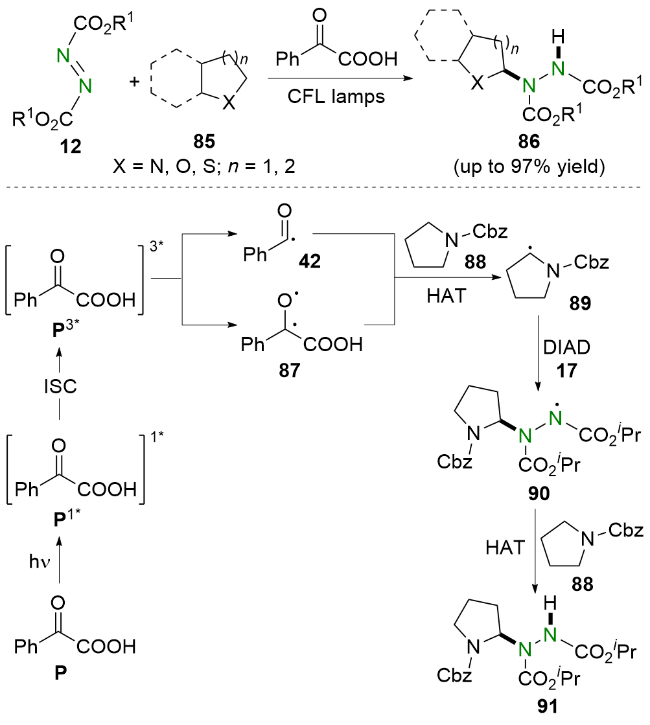
2020年, Kokotos等[37]使用苯甲酰甲酸作为光催化剂, 在家用灯或太阳光的照射下合成了偶氮二羧酸酯的氢烷基化产物(Scheme 17). 值得一提的是, 该策略不仅适用于氮杂环, 而且适用于氧杂环和硫杂环. 相关的机理研究表明, 苯甲酰甲酸P在光照下到达单线态P1*, 并通过系间窜越(ISC)到三线态P3*, 随后产生自由基中间体42和87. 之后, 自由基中间体42和87从N-保护四氢吡咯88中攫取氢原子, 产生α-氨烷基自由基89. 该自由基中间体89则会被DIAD 17捕获, 生成氮中心自由基90. 最后, 自由基中间体90与N-保护四氢吡咯88发生氢原子转移得到目标产物91并再生α-氨烷基自由基89.
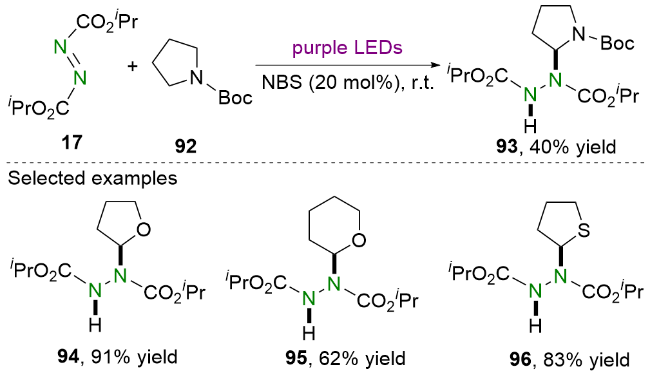
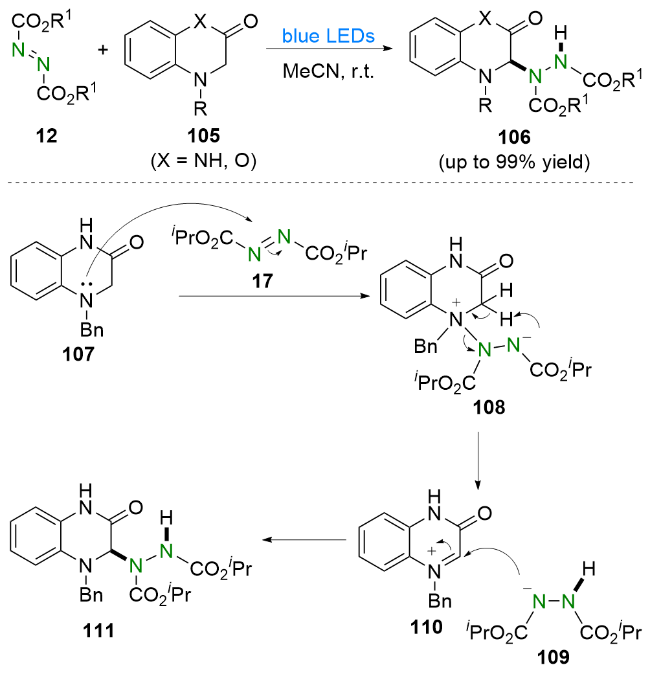
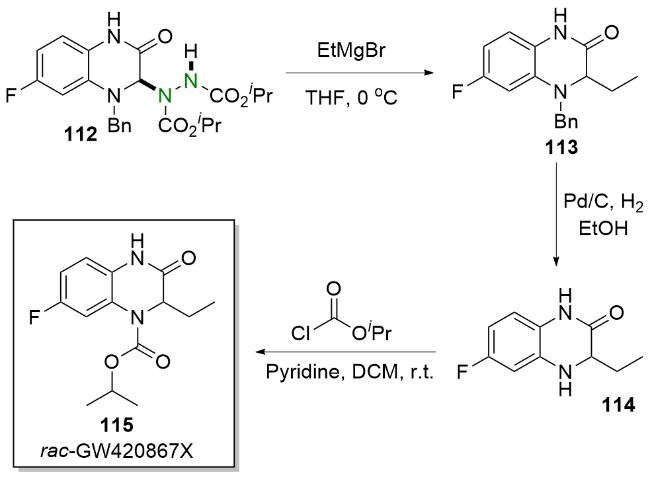
依据实验结果, 他们提出了一个可能的反应机理. 首先, DIAD 17在蓝光的照射下被活化. 之后, 喹喔啉酮107与DIAD 17发生亲核加成生成两性离子中间体108, 并随后产生亚胺离子110和氮阴离子109. 最后, 通过氮阴离子109对亚胺离子110的亲核进攻得到氢烷基化产物111. 值得一提的是, 该方法已被成功应用于生物活性化合物rac-GW420867X (115)的简洁合成(Scheme 21).
2.3 环化反应
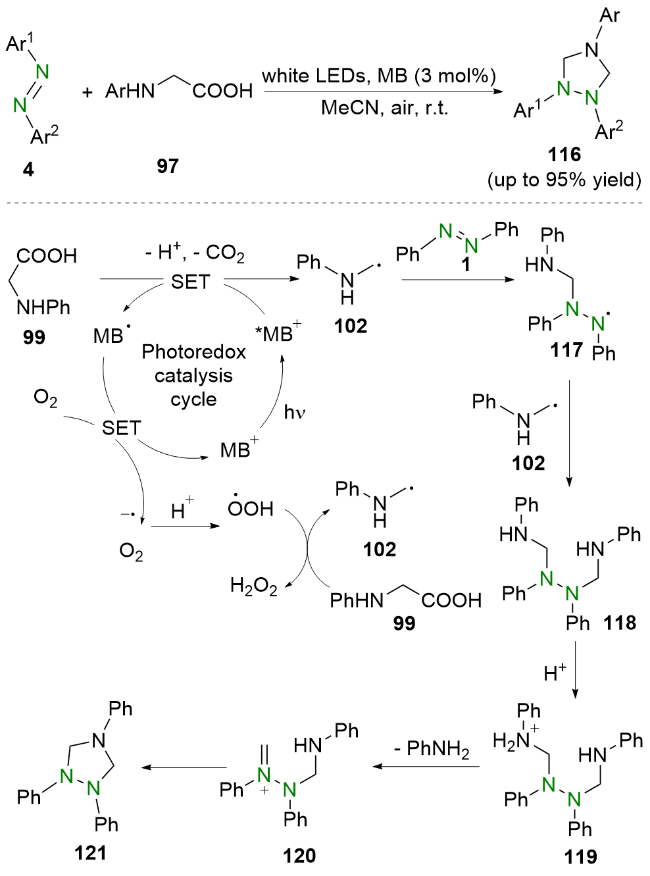
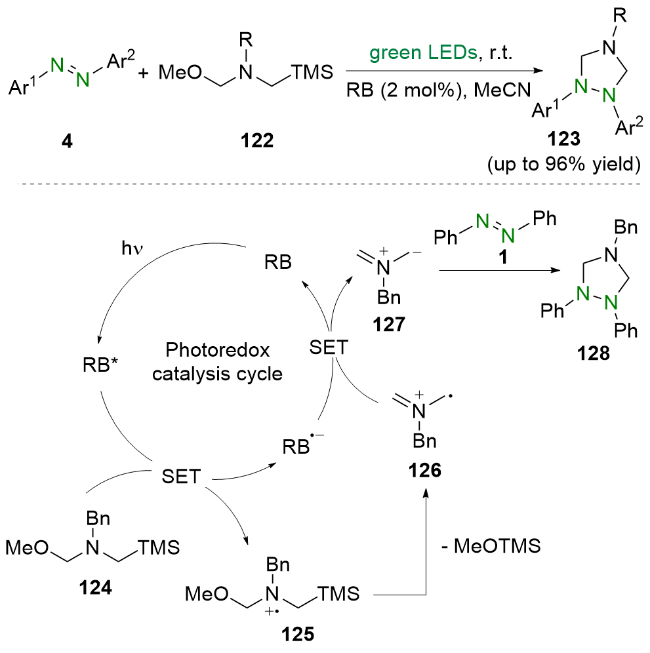
2021年, 周红艳和杨靖亚等[43]报道了可见光驱动的偶氮苯类化合物与甘氨酸衍生物的脱羧加成环化反应(Scheme 22). 他们使用市售的亚甲基蓝作为光催化剂, 在室温下以较高的收率实现了1,2,4-三唑啉衍生物的合成. 值得一提的是, 该工作中合成了此前难以有效合成的1,2-二芳基型1,2,4-三唑啉. 机理研究和文献报道表明, 亚甲基蓝(MB+)在可见光的照射下到达激发态*MB+, 然后与苯甘氨酸99发生单电子转移, 生成α-氨烷基自由基102和亚甲基蓝自由基MB•, 随后, 自由基中间体102与偶氮苯1发生自由基加成生成氮中心自由基117, 该自由基中间体117再与另一分子α-氨烷基自由基102偶联生成中间体118. 之后, 该中间体118经过质子化再脱去一分子苯胺得到氨基阳离子120. 最后, 氨基阳离子120经历分子内环化以及去质子化即可得到环化产物121.
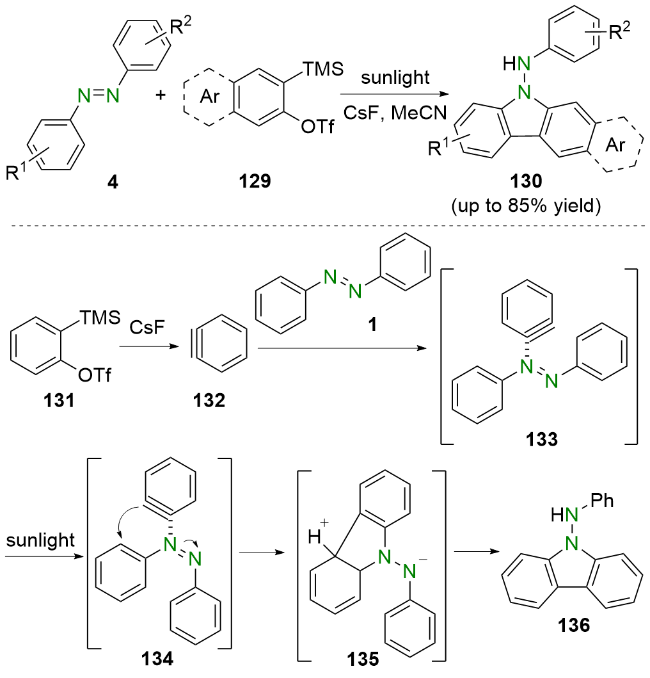
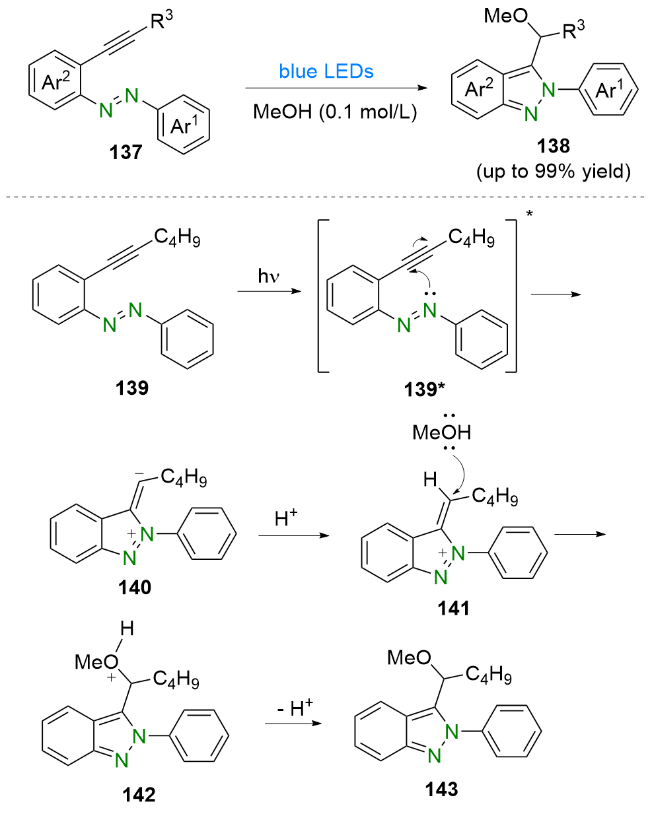
根据理论计算和实验研究的结果, 他们提出了一个可能的反应机理. 首先, 2-(三甲基硅)苯基三氟甲烷磺酸盐(131)在氟化铯的作用下原位生成苯炔132, 然后与偶氮苯1反应生成中间体133. 该中间体133在阳光的照射下转变为中间体134. 随后, 中间体134经过分子内环化生成五元中间体135. 最后, 中间体135在微量水的作用下脱质子芳构化, 同时质子化生成最终产物136.
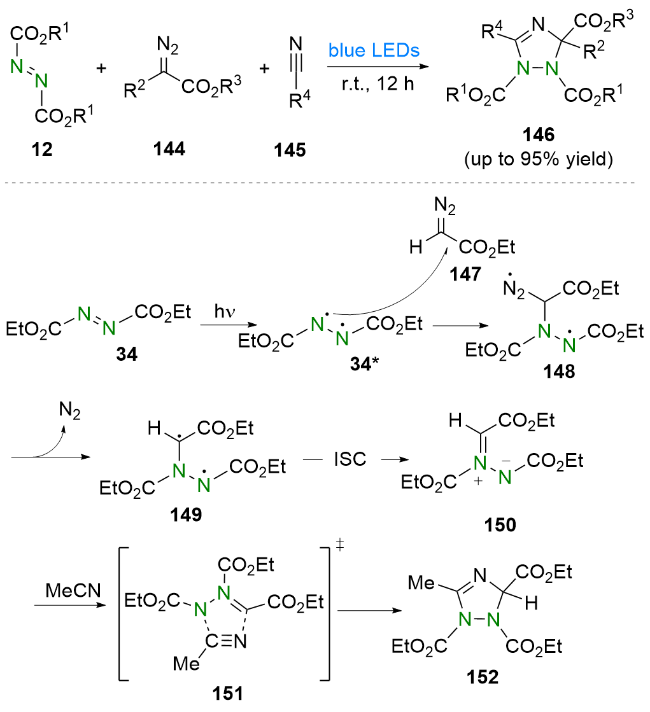
3 其他化学键的构建
3.1 C—C键的构建
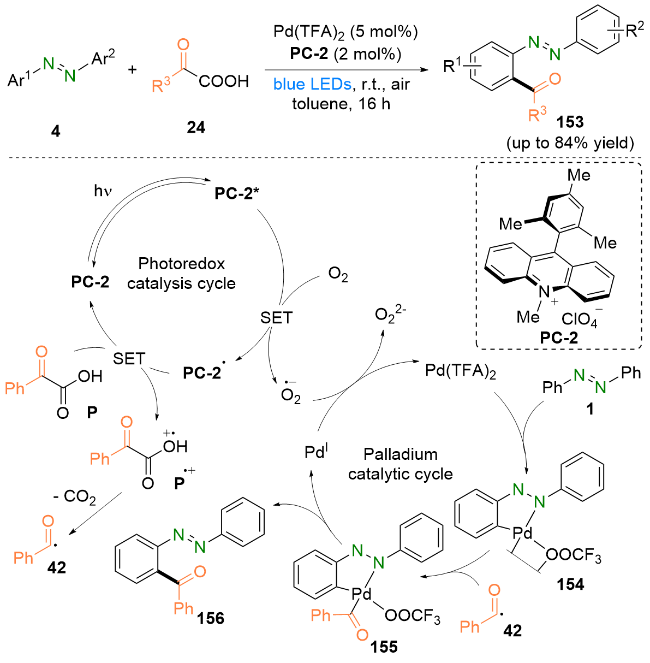
基于实验结果和相关文献报道, 作者提出了一个可能的反应机理. 首先, 光催化剂PC-2在光照下到达激发态PC-2*, 随后被氧气猝灭生成PC-2•和$\mathrm{O}_{2}^{·-}$. PC-2•进而与苯甲酰甲酸P发生单电子转移生成苯甲酰甲酸自由基阳离子P•+, 并再生光催化剂PC-2, 完成光氧化还原催化循环. 之后, 苯甲酰甲酸自由基阳离子P•+脱羧生成酰基自由基42. 另一方面, 偶氮苯1上偶氮基邻位C—H键被Pd(TFA)2活化生成中间体154. 随后捕获酰基自由基42生成中间体155. 之后, 中间体155发生还原消除得到目标产物156, 同时生成PdI. 最后, $\mathrm{O}_{2}^{·-}$氧化PdI得到Pd(TFA)2完成钯催化循环.
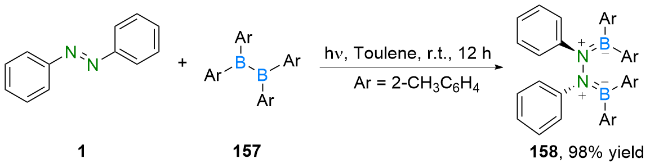
3.2 N—B键的构建
3.3 N—P键的构建
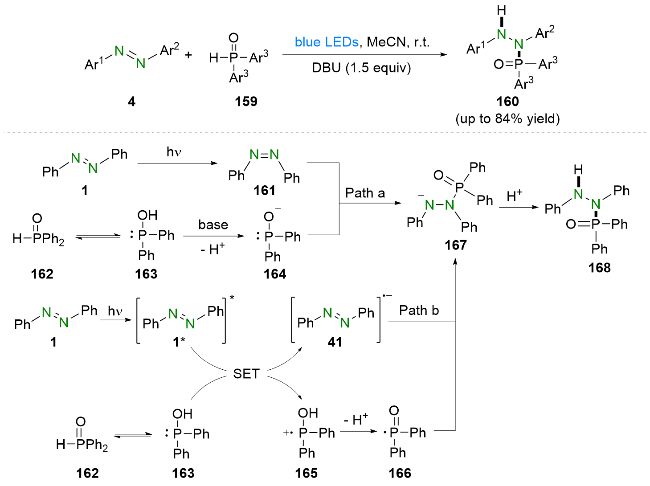
2022年, 周红艳和杨靖亚等[53]报道了可见光驱动的偶氮苯类化合物与二芳基膦氧化物的氢磷化反应(Scheme 29). 值得注意的是, 该反应能够顺利进行的关键之处在于反式偶氮苯会在可见光驱动下异构成为顺式偶氮苯. 此外, 该策略不需要任何光催化剂, 即可在温和的反应条件下构建氮-磷键. 基于实验结果和相关文献报道, 他们提出了如Scheme 29所示的反应机理. 首先, 反式偶氮苯1在可见光照射下经历光异构化得到顺式偶氮苯161. 与此同时, 二苯基氧膦162互变异构成为三价膦163, 并在碱的作用下脱质子生成阴离子中间体164. 随后, 阴离子中间体164进攻顺式偶氮苯161再经历一步质子化即可得到氢磷化产物168. 该反应另一种可能的途径为: 偶氮苯1首先在光照下到达激发态1*, 随后与二苯基氧膦162的互变异构体三价膦163发生单电子转移, 生成偶氮苯自由基阴离子41以及自由基阳离子165. 之后, 自由基阳离子165在碱的作用下脱质子生成磷中心自由基166, 进而与偶氮苯自由基阴离子41偶联得到氮阴离子167. 最后, 氮阴离子中间体167再经过质子化即可得到氢磷化产物168.
3.4 N—N键的构建
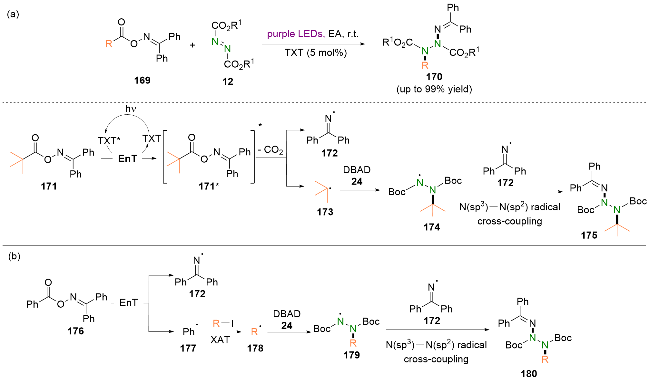
最近, 夏鹏举等[54]使用多种肟酯双官能化试剂作为烷基/苯基/磺酰基和亚胺自由基的来源, 实现了可见光照射下偶氮二羧酸酯的双官能化(Scheme 30, a). 值得一提的是, 氮-氮键的构建依赖于肼自由基和亚胺自由基之间罕见的N(sp3)—N(sp2)自由基偶联. 依据实验结果和相关文献报道, 他们提出了一个可能的反应机理. 首先, 双官能化试剂171通过与激发态的噻吨酮发生能量转移到达激发态171*. 随后, 激发态的双官能化试剂171*发生N—O键均裂并脱羧, 得到叔丁基自由基173和亚胺自由基172. 之后, 叔丁基自由基173被DBAD 24捕获, 生成肼自由基174. 最后, 肼自由基174和亚胺自由基172之间发生自由基交叉偶联生成双官能化产物175.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4 总结与展望
综上所述, 本文总结了近年来可见光促进偶氮化合物参与的光化学转化. 主要包括N—H键、C—N键以及其他类型化学键的构建. 尽管该领域已经取得了较大的进展, 但是仍存在着一些挑战性的问题. 例如, 大多数的方法不适用于非对称的偶氮化合物; 其次, 相较于偶氮二羧酸酯, 偶氮苯类化合物的光化学转化较为欠缺, 而且更具挑战; 再次, 目前已报道的方法大多集中在构建N—H和C—N键, 而其他类型化学键的构建则相对较少; 最后, 对偶氮化合物进行双官能化的反应类型还有待进一步深入研究.
总之, 在可见光促进偶氮化合物参与化学转化这个领域仍存在许多挑战和机遇. 相信通过有机工作者们的持续努力, 该领域定会蓬勃发展, 更多新颖的反应将会在不久的将来陆续出现.
(Zhao, C.)