


1 Introduction
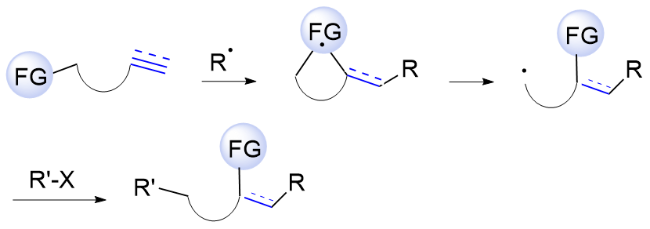
Alkenes and alkynes, prevalent in natural products, serve as key synthetic feedstocks in organic synthesis, medicinal chemistry, and materials science. The difunctionalization of unsaturated hydrocarbons represents one of the most efficient and straightforward methods for their application, enabling the sequential introduction of two new functional groups across the unsaturated bond in a single step.[1] To date, significant progress has been made in recent decades. Notably, visible-light-driven photocatalysis for free radical-induced functional group migration (FGM) stands out for its regioselective difunctionalization of alkenes and alkynes, with promising prospects for future research.[2] Mechanistically, the difunctionalization reaction involves a cascade sequence that begins with radical addition, inter- or intramolecularly, proceeds through cyclic transition states for functional group migration, and culminates in radical quenching to yield the desired products. The reaction efficiency of the cascade hinges on each elementary step, where the electronic properties of the migrating groups are crucial for the migration feasibility. The cleavage of cyclic transition states provides the driving force for functional group migration to facilitate the reaction, as depicted in Scheme 1. Although numerous reviews have documented significant advancements in radical-me- diated functional group migration,[3] there is a clear need to emphasize recent progress in the difunctionalization of unsaturated hydrocarbons. This emphasis is particularly important for developments involving functional group migration strategies under photo-driven conditions, especially considering the rapid evolution of photocatalysis over recent decades. This review aims to highlight recent progress in the photocatalytic difunctionalization of unsaturated hydrocarbons through radical-mediated functional group migration. The discussion is organized into three sections that elaborate on distinct approaches to difunctionalization: aryl group migrations across diverse olefinic substrate skeletons, bifunctional reagents mediated difunctionalization via functional group migration, and other functional group migration pathways for the difunctionalization of unsaturated hydrocarbons.
2 Aryl group migration across various olefinic substrate skeletons
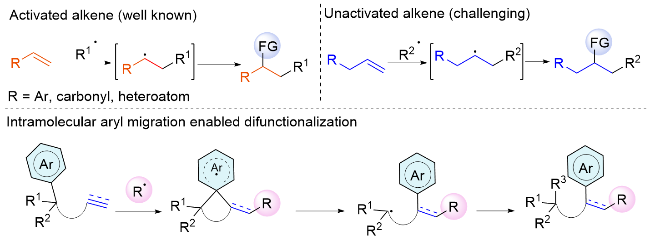
Visible light-mediated difunctionalization of alkenes stands as a potent strategy for augmenting molecular diversity and complexity.[4] In recent decades, significant strides have been made within this field. Nonetheless, most studies have concentrated on activated alkenes with functional groups including aryl, carbonyl, and heteroatoms (Scheme 2). The presence of an adjacent π-system, with its low-lying lowest unoccupied molecular orbital (LUMO) energy, promotes interaction with the single occupied molecular orbital (SOMO) of the nascent radicals, leading to the stabilization of these radicals through π-π conjugation or π-orbital delocalization. In contrast, non-activated alkenes exhibit polarity mismatches that render certain nucleophilic radicals less reactive towards electron-rich double bonds, thereby restricting the range of radical precursors. Furthermore, the nucleophilic radicals produced by the addition to non-activated alkenes are ephemeral and highly unstable. In this scenario, intramolecular functional group migration emerges as a privileged tactic to treat unactivated alkenes. Furthermore, a variety of distal olefinic substrate framworks have been designed and utilized as models for the radical difunctionalization.
2.1 Distal tertiary alcohol substituted alkenes
Tertiary alcohol-substituted alkenes are among the most extensively studied substrates for radical addition and aryl migration cascades, facilitating the difunctionalization of alkenes.[5] Under photocatalytic conditions, electron transfer from the excited photocatalyst to the radical precursor generates radical 2-1. Subsequently, the radical attacks the alkene, yielding the nascent alkyl radical 1-1, which subsequently attacks the aromatic ring, leading to the formation of a cyclic transition state 1-2. This triggers the homolytic cleavage of the carbon-carbon bond, resulting in aryl migration. Ultimately, oxidation of the carbon-cen- tered radical leads to the formation of difunctionalized ketone products (Scheme 3).
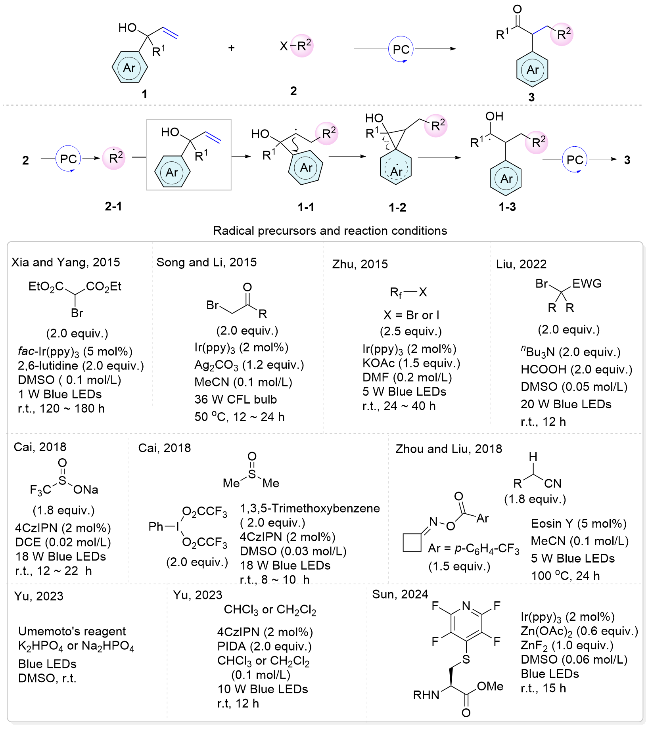
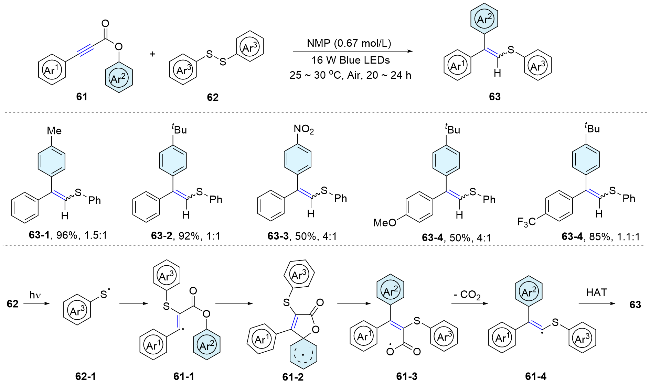
Numerous radical precursors have been examined for the difunctionalization of alkenes with distal tertiary alcohols. Alkyl halide has proved to be an efficient radical precursor under photoredox conditions. In 2015, Xia and Yang’s group[6] described the 1,2-aryl migration of allyl alcohols under blue light irradiation, affording a range of arylalkylated alkenes. Notably, this reaction yields two regioisomeric products when the allyl alcohol substrate bears two distinct aryl groups. Typically, electron-deficient aryl groups migrate more readily than those that are electron-rich. However, the reaction was hampered by prolonged reaction times, excessive photosensitizer requirements, and substrate scope limitations. Subsequently, Song and Li et al.[7] presented a method that addressed these issues. In the same year, Zhu’s group[8] integrated a fluoroalkyl moiety into the system, enabling arylfluoroalkylation of alkenes. To get rid of the costly Ir(ppy)3 photocatalyst, our group recently achieved a photocatalyst-free procedure by forming an electron-donor-acceptor (EDA) complex between Bu3N and alkyl bromides to realize this radical addition/aryl migration cascade reactions under visible light irradiation (Scheme 3).[9]
Various other radical precursors have been demonstrated to effectively facilitate 1,2-aryl migration reactions in allyl alcohol substrates.[10] In 2017, Zhu and co-workers reported the first chemo- and regioselective distal heteroaryl ipso-migration for the difunctionalization of unactivated alkenes in the presence of hypervalent iodine reagent. A variety of fluoroalkyl functionalized heteroarenes could be prepared under mild reaction conditions.[11] In 2018, Cai’s group[12] employed sodium trifluoromethanesulfinate as a radical precursor that, upon single-electron oxidation, releases sulfur dioxide and forms a trifluoromethyl radical, facilitating aryl trifluoromethylation of alkenes. Furthermore, under photoredox catalysis conditions, Umemoto’s reagent can also be applied in this transformation.[13] In the same year, Cai’s group[14] presented a metal-free oxidative 1,2-alkylarylation of unactivated alkenes using dimethyl sulfoxide. The in situ generated hypervalent iodine reagent was identified as a key hydrogen atom transfer (HAT) precursor, yielding the active α-sulfinyl radical and facilitating the reaction. In 2018, Zhou and Liu’s group[15] employed the ring-opening of a cyclobutanone oxime ester to generate an alkyl radical as a HAT agent to abstract a hydrogen atom from the α-carbon of a nitrile group, thus creating an electrophilic radical for the reaction. In 2023, Yu’s group[16] successfully introduced alkyl chloride moieties into the reaction using chloroform or dichloromethane as radical precursors. Recently, Sun and co-workers[17] utilized the excited-state photosensitizer to activate the thiol group in cysteine derivatives, generating carbon-centered radical intermediates through desulfurization to participate in this cascade radical difunctionalization reactions.
Additionally, elongation of the carbon chain between the tertiary alcohol moiety and double bond enables smooth 1,3- or 1,4-aryl migration. In 2017, the Gu group[18] employed sulfate radical anion, generated from persulfate, as an HAT reagent to activate the C—H bond adjacent to oxygen, initiating the radical addition and 1,4-aryl migration (Scheme 4, A). Subsequently, Pan and co-workers[19] developed N-hydroxybenzimidoylchloride (NHBC) ester as fluoroalkyl radical precursors for the efficient fluoroalkylheteroarylation of unactivated olefins (Scheme 4, B). Similar results were independently achieved by the groups of Ngai and Ji, utilizing aryl acyl chlorides as radical precursors.[20,21] In the same year, Zhu’s research group[22] applied this strategy to the difunctionalization of alkynes, synthesizing a variety of all-carbon-tetrasubstituted olefins with moderate to good yields and stereoselectivity (Scheme 4, C).
In 2017, the Zhu’s group[23] described an efficient radical fluoroalkyl heteroarylation of unactivated alkenes via distal ipso-migration of heteroaryls under photoredox catalysis (Scheme 5). Notably, the migratory aptitude of different heteroaryls was comprehensively investigated by comparing the migration ratios under the standard conditions. Among them, benzofuran demonstrated the highest migratory propensity, followed by furan and thiazole with similar abilities, and thiophene with the lowest capability. These differences were partially attributed to discrepancies in the energy of the LUMO levels among the corresponding heteroaryl groups.
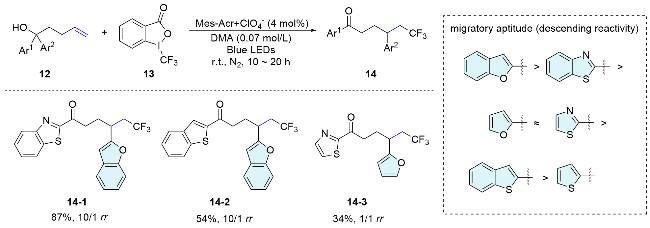
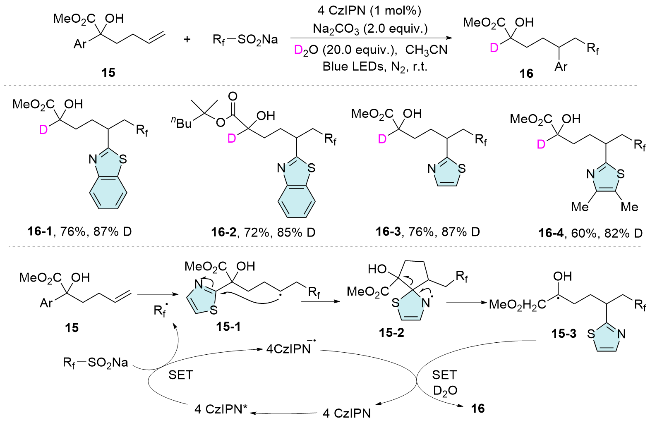
In early 2024, Zhu and co-workers[24] reported a novel and efficient photoredox-neutral 1,2,5-trifunctionalization of α-hydroxyhexenoates through a cascade reaction involving radical addition, heteroaryl migration, and radical-polar crossover deuteration. This method provides an efficient approach for the synthesis of valuable α-deute- rated lactic acid derivatives, with a wide range of functional groups being well tolerated under mild reaction conditions. Mechanistically, the excited photocatalyst undergoes single electron oxidation of CF3SO2Na to generate a fluoroalkyl radical, which adds to the alkene to form alkyl radical 15-1. Subsequently, the alkyl radical attacks the heteroarene, leading to the formation of the spiro radical intermediate 15-2. The homolytic cleavage of the C—C bond, followed by single electron reduction and deuteration, yields the trifunctionalized product 16 (Scheme 6).
2.2 Distal sulfonamide bearing alkenes
The distal aryl sulfonamide-substituted alkene framework has also proven to be a representative scaffold for radical addition and aryl migration. Several seminal works on classical radical Smiles rearrangement reactions have been reported by the Nevado’s group[25] within transition metal catalysis. More recently, this framework has been integrated into photoredox catalytic systems, enabling diverse difunctionalization of alkenes. Mechanistically, the reaction initiates with the formation of the radical intermediate 18-1 through single-electron transfer between radical precursors and excited photocatalysts. Subsequently, the radical adds to the double bond of the alkene, generating a nascent radical 17-1, which then attacks the aryl sulfonyl group to form radical 17-2. Then, homolytic cleavage of the C—S bond releases SO2, yielding a nitrogen-centered radical 17-3. Finally, the radical cyclization followed by oxidative rearomatization leads to the formation of compound 19, whereas hydrogen atom transfer (HAT) yields the corresponding amide 20 (Scheme 7).
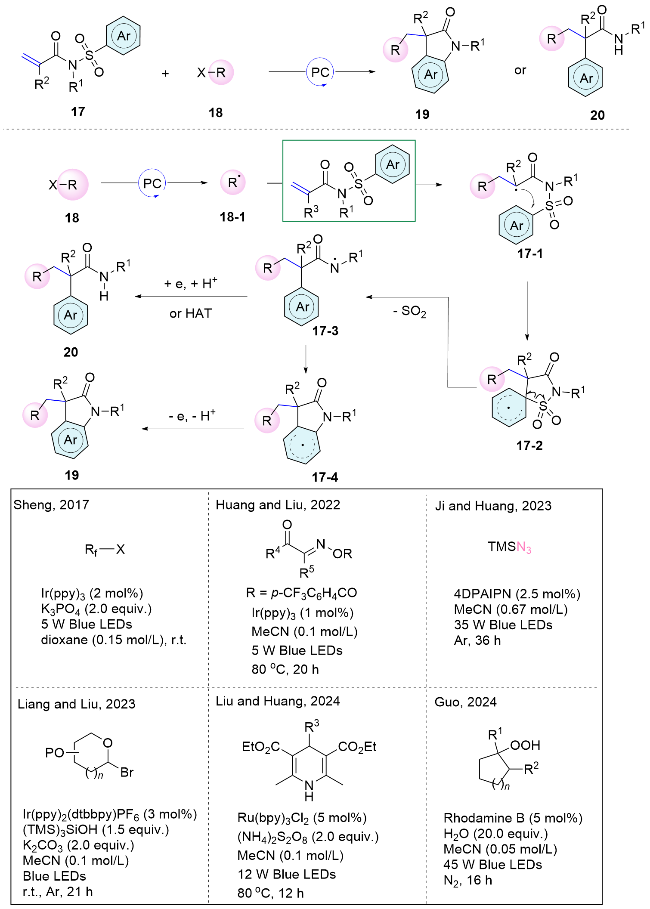
In 2017, Sheng and co-workers[26] employed fluoroalkyl iodides as radical precursors, enabling the generation of fluoroalkyl radicals via single-electron reduction by an excited photocatalyst, thereby initiating the difunctionalization reaction. Interestingly, the selective synthesis of amide product 20 or cyclized oxindoles 19 can be achieved by altering the substituents on the nitrogen atom of the tosyl amides. In 2022, Huang, Liu, and colleagues[27] reported a cascade process that encompasses acyl radical addition to carbon-carbon double bonds, followed by aryl migration, desulfurization, and intramolecular cyclization. This redox-neutral approach exhibits remarkable selectivity, compatibility with diverse substituents, and a wide range of substrate scope. In 2023, Ji’s group[28] employed trimethylsilyl azide (TMSN3) as the azide radical precursor under photocatalyzed conditions. Mechanistic investigations indicate that the initial step comprises single-electron transfer (SET) between TMSN3 and the excited photocatalyst, which is succeeded by radical addition, aryl migration, and desulfurization, culminating in the synthesis of α-aryl-β-azidoamides and azidoxindoles under mild conditions. Liang and Liu[29] employed (TMS)3SiOH as the XAT reagent to initiate the glycoarylation of activated olefins with glycosyl bromides. Recently, Liu and Huang[30] further employed Hantzsch esters as alkyl radical precursors to capture sulfur dioxide generating sulfinyl radicals to promote the corresponding cascade difunctionalization reactions. The Guo’s group[31] recently reported the cleavage of C—C bonds in peroxides to generate alkyl radicals to accomplish Smiles rearrangement mediated difunctionalization within this class of amide frameworks (Scheme 7).
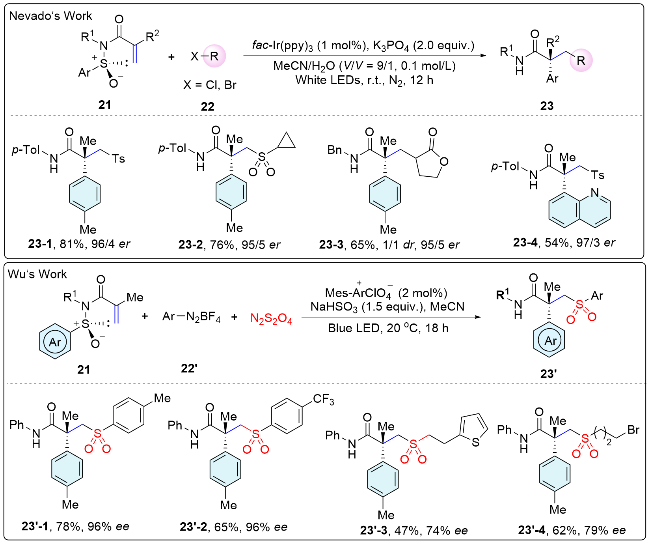
In 2021, the Nevado group[32] for the first time utilized enantioenriched N-aryl sulfinyl acrylamide as acceptors for a variety of radicals produced under mild photoredox conditions to access acyclic amides that bear a chiral α-all-carbon quaternary center (Scheme 8). The sulfinamido group not only directs the 1,4-migration of the aryl moiety onto the α-carbon of the amide, which thus governs its absolute configuration, but also functions as a traceless chiral auxiliary. The method features mild reaction conditions, broad substrate scope and excellent enantioselectivity. Recently, a similar strategy was reported by Wu and co-workers.[33] They present two complementary photoinduced sulfur dioxide insertion systems to trigger radical asymmetric Truce-Smiles rearrangements for preparing a variety of chiral sulfones that bear a quaternary carbon stereocenter (Scheme 8).
In 2021, a radical triggered fragmentary cyclization cascade reaction of ene-ynamides was presented by Shu and co-workers,[34] providing a rapid access into [1,2]-annulated indoles by an intermolecular radical addition, intramolecular cyclization, desulfonylative aryl migration, and site- selective C(sp2)—N cyclization sequence. More diverse [1,2]-annulated indole skeletons can also be prepared by this approach. Density functional theory (DFT) calculations revealed that an aza-Nazarov type cyclization is responsible for the excellent site-selectivity observed in the C(sp2)—N bond forming step (Scheme 9).
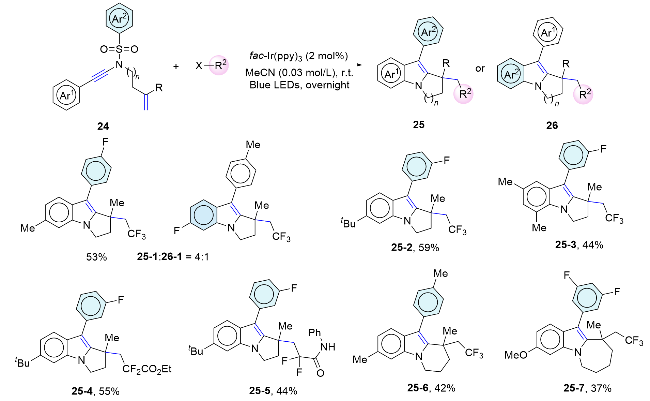
In 2022, Stephenson’s group[35] disclose an intramolecular alkene aminoarylation cascade reaction for unactivated alkenes. This method is compatible with a wide range of unactivated alkenes, which is distinct from current cross-coupling techniques, and facilitates the synthesis of highly substituted arylethylamines using readily available aryl sulfonamides and unactivated alkenes under mild conditions. A stepwise photoredox mechanism was proposed. Upon photoexcitation, Ir(III)* oxidizes the deprotonated N-acylsulfonamide (27-1) to generate the N-centered radical (27-2). Subsequently, the C—N bond formation occurs through a 5-exo-trig cyclization, leading to the formation of an alkyl radical (27-3) that reacts with an arene to form a dearomatized spirocyclic intermediate (27-4). The subsequent desulfonylation, followed by reduction with iridium(II), regenerates the photocatalyst and yields an amidyl anion (27-5). This anion then irreversibly deprotonates either benzoic acid or an additional equivalent of the starting material to afford the final product (Scheme 10).
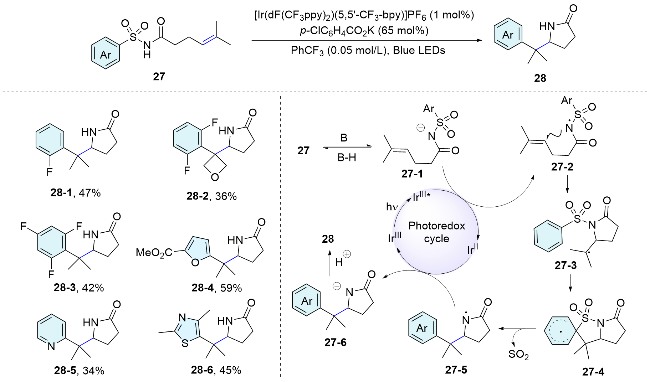
In addition, the sulfonamide framework can also be used to trigger the difunctionalization of alkynes. In 2016, the Xia’s group[36] disclosed that linear 3-aryl-N-(arylsulfonyl)-propiolamides underwent an intramolecular cascade cyclization reaction for the synthesis of phenanthrene derivatives under ultraviolet irradiation. Mechanistically, the alkyne moiety in the substrate is excited into a 1,2-biradi- cal species 29-1, which then initiates a tandem Smiles rearrangement to afford biradical 29-2. Sequential C—S bonding via 1,5-biradical cyclization affords the five- membered isothiazol-3(2H)-one 1,1-dioxide 30. Finally, the typical Mallory reaction provides phenanthrene derivative 31. The control experiment results and isolation of the key intermediates 30 give further insight into the reaction mechanism (Scheme 11).
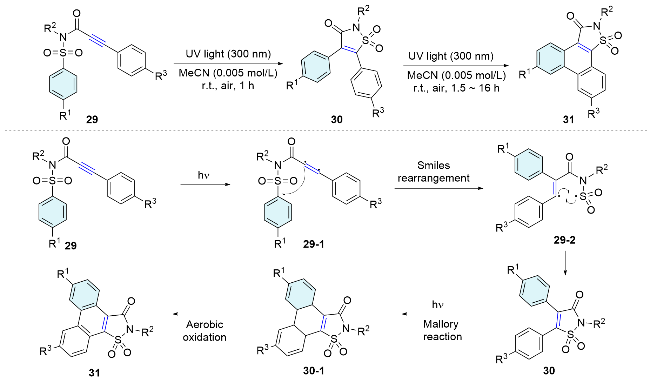
Polycyclic indole scaffolds are ubiquitous in pharmaceuticals and natural products, and in materials science. In 2020, Zhang and co-workers[37] present a visible-light-induced intramolecular aryl migration/desulfonylation/cyclization cascade process for the synthesis of tetracyclic indolo[2,1-a]isoquinolin-6(5H)-ones from distal alkyl bro- mide substituted alkynes under mild conditions (Scheme 12, A). Moreover, 1,n-enynes are privileged building blocks for radical cascade cyclization as highly complex structures of chemical and biomedical importance can be generated through the synergistic cascade processes across C=C and C≡C bonds. In 2017, Li and co-workers[38] disclosed a visible-light-mediated radical cascade reaction of 1,8-enynes to synthesize a wide range of difluoroalkylated tetracycles with indole and dihydroquinolinone scaffolds. Notably, four new chemical bonds are formed in one single operation. A cascade process involving radical addition, Smiles rearrangement, radical cyclization and oxidative radical cyclization was involved (Scheme 12, B).
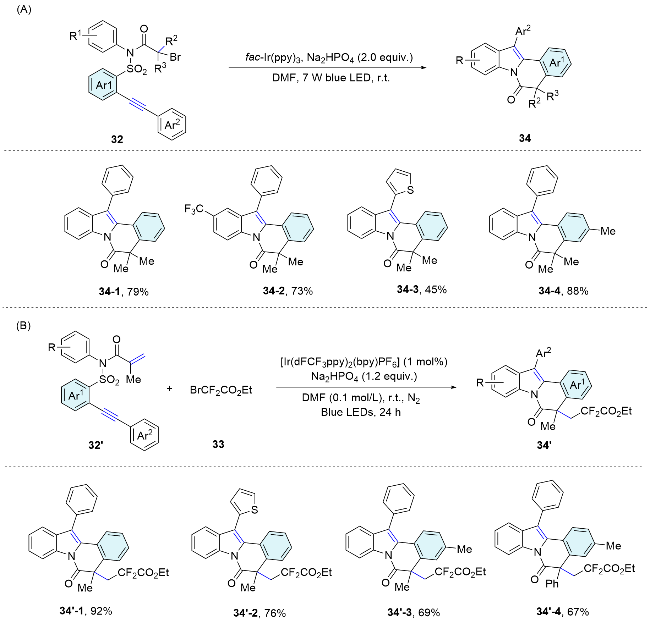
In 2020, Wu’s group[39] developed an efficient radical Smiles rearrangement initiated by neutral eosin Y based HAT photocatalysis. The transformation proceeds via a cascade sequence involving visible-light-induced HAT, 1,4-addition, Smiles rearrangement, 5-endo-trig cyclization, and a reverse HAT process. The merits of this transformation include atom and step economy, the absence of metal and additive requirements, excellent selectivity, and the facile generation of heterocyclic scaffolds that are otherwise challenging to synthesize. This method, applicable to a variety of aldehydes and a broad scope of sulfonamide substrates, enables the efficient synthesis of highly functionalized isothiazolidinone compounds. These compounds are of potential biological interest (Scheme 13).
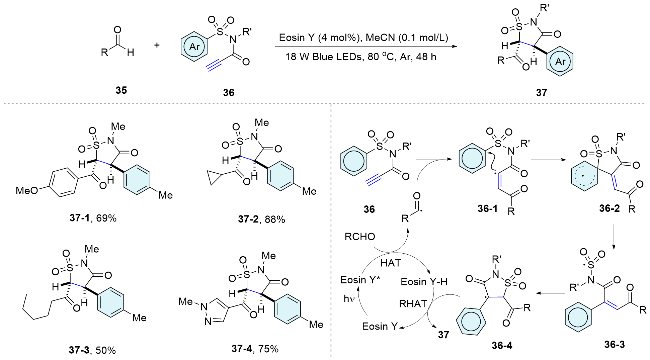
2.3 Distal pyridinium substituted alkenes
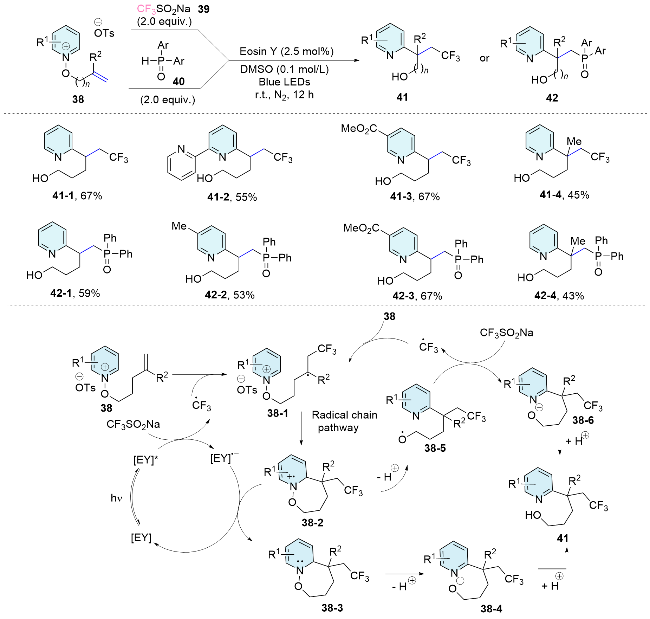
Pyridine compounds, ubiquitous in natural products and pharmaceuticals, have motivated sustained research for their targeted functionalization, with particular emphasis on the C2 and C4 positions, highlighting their critical role in structural diversity and biological activity.[40] Distal pyridinium-substituted alkenes have become a valuable framework for the heteroarylation of unactivated alkenes through an intramolecular pyridyl shift. In 2020, Hong’s team[41] described a visible-light-mediated, site-selective trifluoromethylpyridylation of alkenes, realized through a remote pyridyl ortho-selective migration, yielding synthetically valuable pyridines with C2-fluoroalkyl functionalities. Two plausible mechanistic pathways have been posited. In the photoredox catalytic cycle, the photoexcited state EY* is subjected to single-electron oxidation by the Langlois reagent, producing an electrophilic CF3 radical that reacts with the alkene, yielding intermediate 38-1. Subsequently, the nucleophilic alkyl radical undergoes intramolecular addition to the C2-position of the pyridinium substrate, forming the radical cation intermediate 38-2, which is then reduced by the photocatalyst to afford 38-3. Ultimately, deprotonation and subsequent protonation result in the formation of the difunctionalized product 41. Alternatively, a radical chain mechanism may be implicated, with intermediate 38-2 undergoing deprotonation and subsequent N—O bond cleavage, generating alkoxy radicals. Subsequently, a single-electron transfer occurs between 38-5 and CF3SO2Na, initiating the radical chain that leads to the functionalized alcohol upon protonation. The relatively high quantum yield of the reaction supports that both mechanistic pathways were involved in the cascade reaction. Notably, this approach has been successfully broadened to the reaction of phosphorus-centered radicals, facilitating the synthesis of quaternary carbon centers bearing ortho-substituted pyridyl groups while retaining excellent C2 selectivity (Scheme 14).
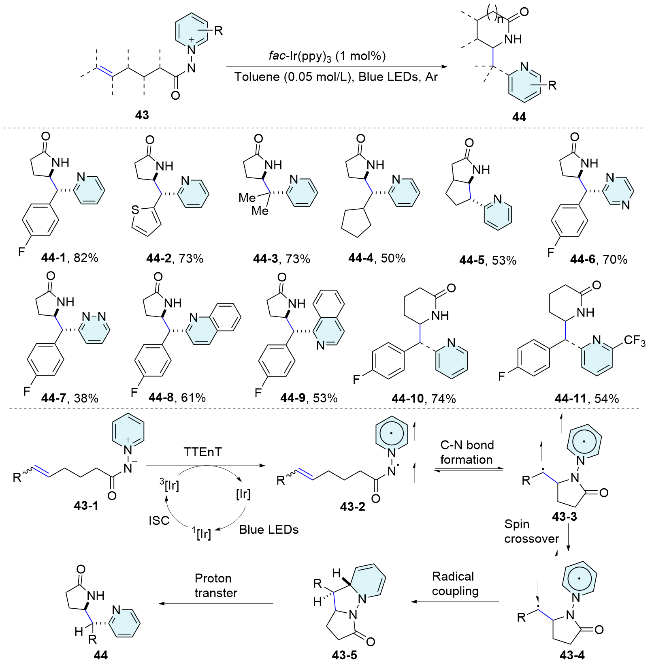
In 2023, the same group[42] described an efficient approach for the diastereoselective pyridyl lactamization via a photoinduced [3+2] cycloaddition (Scheme 15). This method demonstrates good efficiency, diastereoselectivity, and tolerance towards different functional groups, offering a valuable synthon for constructing ortho-pyridyl γ- and δ-lactam scaffolds with a syn-configuration in a single step. A plausible stepwise reaction mechanism involving triplet-triplet energy transfer (TTEnT) from the photocatalyst to the N—N pyridinium ylide 43-1 has been proposed. Upon visible-light irradiation, EnT from photoexcited fac-Ir(ppy)3 to 43-1 results in the formation of the triplet state of the pyridinium ylide 43-2, with an electron transfer energy (ET) of 208.1 kJ/mol. The generated amidyl radical reacts with the tethered olefin on the triplet surface, forming a C—N bond and yielding diradical 43-3. This 1,5- diradical 43-3 undergoes intersystem crossing, followed by recombination of the radicals at the ortho-position of the pyridinium segment, yielding a dearomatized intermediate 43-5. Finally, proton transfer leads to the formation of the pyridyl lactam. Regarding the origin of the observed diastereoselectivity, it is proposed that steric repulsion between substituents on the olefin is a key determinant in the ring-closing radical-radical recombination step.
2.4 Other distal substituted unsaturated hydrocarbons
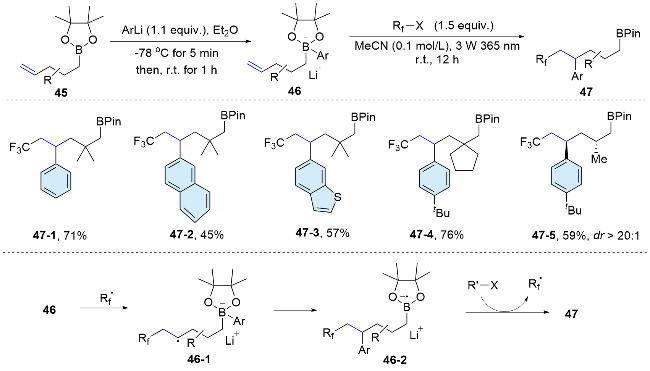
This radical-mediated aryl migration strategy for the difunctionalization of alkenes is not only applicable for the migration of aryl groups from a carbon or heteroatom, but also suitable for the radical aryl migration from a boron center. In 2021, Studer’s group[43] reported a radical 1,5-aryl migration from boron to carbon in aryl boronate com-plexes, achieving arylfluoroalkylation of non-activated alkenes. Upon ultraviolet irradiation, organohalides undergo homolytic cleavage to generate fluoroalkyl radicals, which add to alkenyl aryl boronate complexes to form the radical anion 46-1. Then, the C-radicals engage in 1,5-aryl migration reactions to yield the boronate complex 46-2. Finally, single-electron transfer to perfluoroiodide yields 4-aryl-alkylboronic esters 47. Since boronate complexes can be generated in situ from alkenylboronic acid esters and aryllithium reagents, the aryl group can be readily varied, enabling access to a range of arylated products from a single alkenylboronic acid ester through divergent chemistry (Scheme 16).
In 2021, Shi’s group[44] described that a class of olefins with adjacent all-carbon quaternary centers could undergo 1,2-aryl migration after radical addition. Mechanistically, photoexcitation of the Ir(III) complex with visible light generates a redox-active excited state Ir(III)*. This excited state undergoes single electron transfer (SET) with alkyl halides, leading to the formation of an Ir(IV) species and an alkyl radical. Subsequently, the alkyl radical adds to the alkene, resulting in alkyl radical 49-1. This initiates a 1,2-aryl migration of a vicinal aryl group from the adjacent quaternary carbon center. The resultant Ir(IV) species subsequently oxidizes a tertiary amine, thereby regenerating the Ir(III) complex and producing an amine radical cation A. This radical cation abstracts a hydrogen atom from the trifluoroethanol (TFE) solvent, yielding a carbon-centered radical B. The 1,2-aryl migration generates a C-radical, 49-2, which then abstracts a hydrogen atom from B to complete the domino sequence, ultimately forming the aryl-migrated product and CF3CHO. Notably, the formation of trifluoroethoxy hemiacetal was observed by NMR in the reaction mixture, which is presumed to arise from the reaction of CF3CHO with TFE. In general, the presence of a quaternary carbon with two electron-with- drawing groups is essential for the reaction, which constrains the substrate scope of this method (Scheme 17).
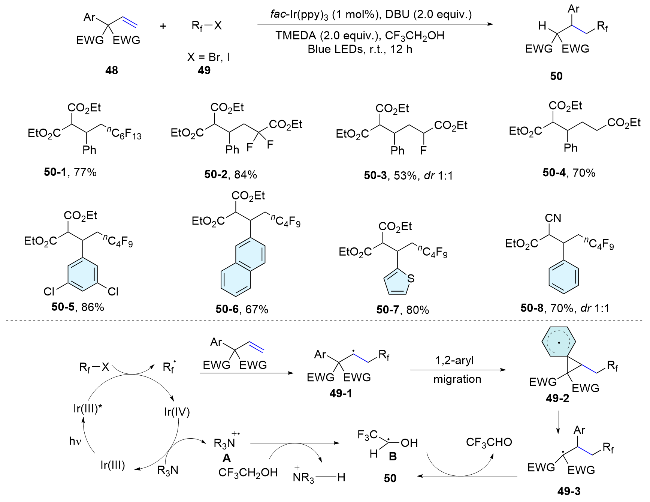
Wang’s group[45] employed the strategy of radical addition and aryl migration for the difunctionalization of non-activated alkenes with distal aryl sulfonyl groups. The reaction is initiated with the oxidation of CF3SO2Na or ArSO2Na by the excited photocatalyst, yielding a trifluoromethyl or sulfonyl radical. The radical reacts with alkene 51 to generate a new alkyl radical 51-1, which then undergoes intramolecular aryl migration to form intermediate 51-3. This intermediate undergoes a single-electron-trans- fer reaction with the reduced photocatalyst, yielding the target product and closing the catalytic cycle. It is noteworthy that this reaction offers an atom-economical protocol for Smiles rearrangements, eliminating the release of SO2 and thereby enabling access to a series of valuable organosulfur products (Scheme 18).
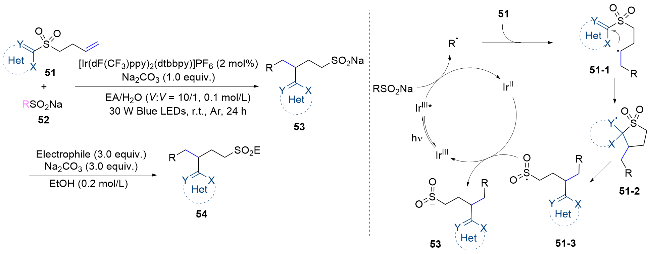
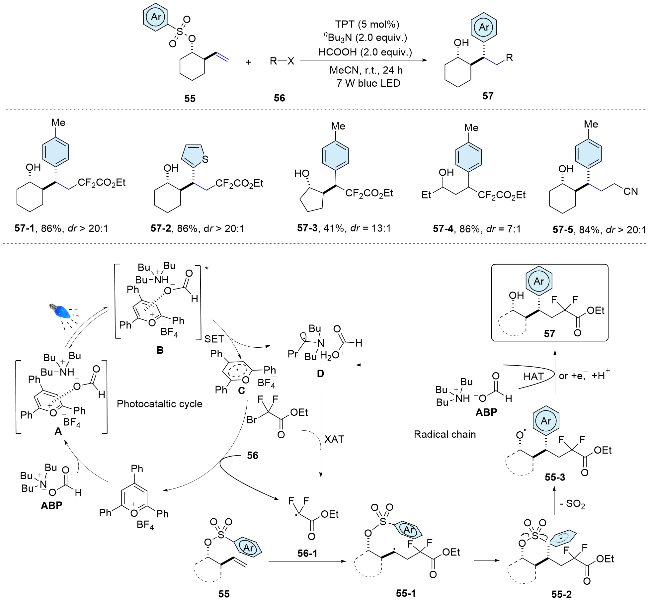
In 2022, our research group[46] achieved fluoroalkyl arylation of non-activated alkenes on distal olefinic aryl sulfonate skeletons. The difunctionalization reaction features a cascade process incorporating radical addition and Smiles rearrangement. This method offers a novel route to tertiary alcohol derivatives with fluoroalkyl moieties, chara- cterized by mild conditions, excellent functional group tole- rance, cost-effective starting materials, and high diastereo- selectivity. Mechanistically, anion-π interactions facilitate the formation of the EDA complex A between the acid base pair (ABP) and the photocatalyst 2,4,6-triphenylpyrylium- fluoroborate (TPT). Subsequently, single-electron transfer from the ABP to the excited B results in the formation of the reductively quenched TPT C and the radical species D. Both intermediates C and D can react with 56 to generate the key fluoroalkyl radical 56-1, either through a SET event or a halogen abstraction process. Thereafter, the electrophilic radical 56-1 adds to the double bond of the sulfonate 55, forming alkyl radical 55-1, which then undergoes a radical Smiles rearrangement to yield alkoxyl radical 55-3. Finally, radical species 55-3 either abstracts a hydrogen atom from the ABP, yielding the desired product and regenerating intermediate D for the radical chain process, or it is reduced by C and subsequently protonated to form the target molecule (Scheme 19).
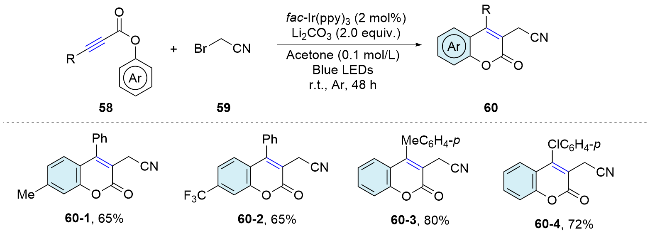
In 2018, Li’s group[47] developed a visible-light-pro- moted method for the mild and direct cyanomethylation of aryl alkynoates (Scheme 20). This method offers a novel alternative for synthesizing 3-cyanomethylated coumarins through a domino sequence involving radical addition, 5-exo cyclization, and ester migration under mild conditions.
Recently, a visible light-promoted radical cascade reaction between alkyl esters and phenyl disulfides at room temperature was reported by the Xiong’s group.[48] The method features additive- and photocatalysts-free. A sequential photolytic cascade reaction involving cleavage of the S—S bond, sulfur radical addition, vinyl radical induced aryl migration, decarboxylation, and hydrogen atom transfer (HAT) afford a series of trisubstituted 1,1-diaryl ethylene sulfides in moderate to good yields (Scheme 21).
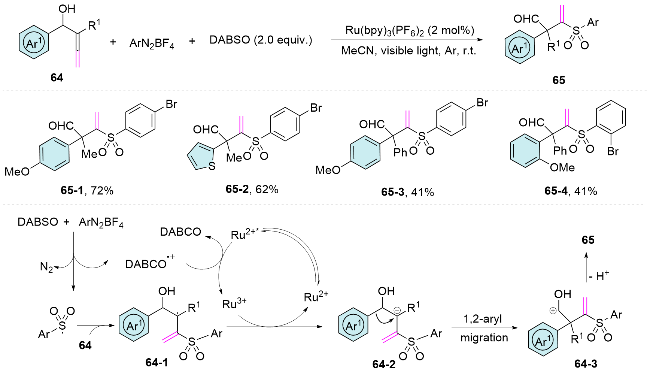
In addition, the strategy of radical addition-induced aryl migration is applicable to the difunctionalization of α-allenols. Almendros and colleagues[49] have introduced an efficient method for synthesizing 2,2-disubstituted 3-(arylsulfonyl)but-3-enals through a three-component coupling reaction of α-allenols, the sulfur dioxide surrogate 1,4-diazabicyclo[2.2.2]octane-1,4-diium-1,4-disulfinate (DABSO), and arenediazonium salts under photoredox conditions. The reaction is proposed to proceed through a sulfonylation-rearrangement cascade. Initially, an electron transfer occurs between DABSO and arenediazonium salts, generating the 1,4-diazabicyclo[2.2.2]octanyl cation radical and the arylsulfonyl radical. These key reactive intermediates then engage with the α-allenol to form allylic radicals, designated as 64-1. Following this, a single-elec- tron oxidation mediated by the photocatalyst leads to the formation of allylic cations, denoted as 64-2. Subsequently, a 1,2-aryl migration step facilitates the formation of cationic homoallylic alcohols, which readily deprotonate to yield the final products, 2,2-disubstituted-3-(arylsulfo- nyl)but-3-enals 65 (Scheme 22).
3 Bifunctional reagent mediated difunctionalization via functional group migration
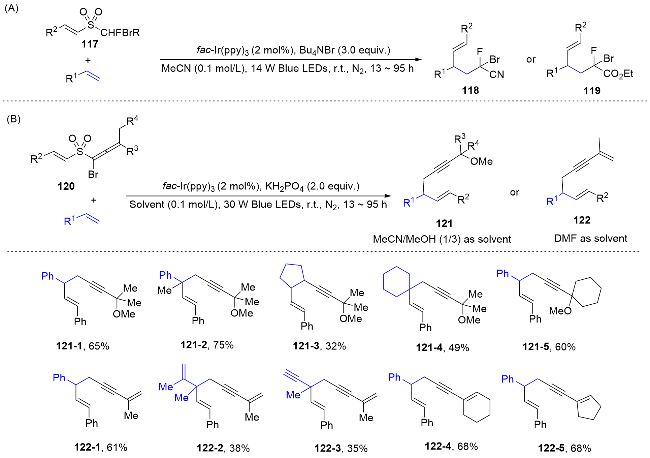
Bifunctional reagents, possessing two reactive sites and capable of being incorporated into unsaturated substrate molecules via cascade reactions have garnered significant attention from the scientific community in recent decades. Numerous examples of bifunctional reagents mediated difunctionalization of alkenes have been extensively documented.[50] The radical docking-migration strategy, pioneered by Zhu and co-workers,[5] is considered a powerful and straightforward method for bifunctional reagent-me- diated difunctionalization. The bifunctional reagent can be efficiently initiated through a single-electron transfer event, yielding reactive radical species. Subsequently, it docks to the alkene or alkynes, followed by intramolecular functional group migration, accomplishing the difunctionalization process. Compared to the above mentioned FGM protocols, the scope of alkenes is significantly expended (Scheme 23).
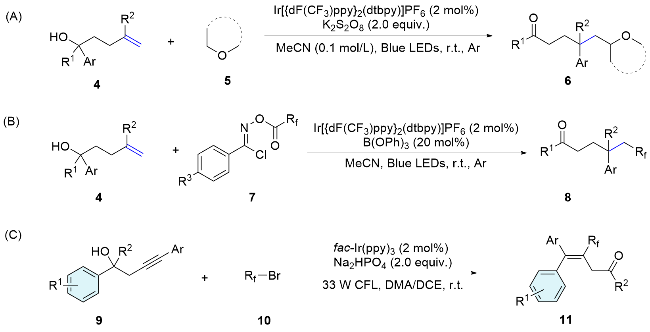
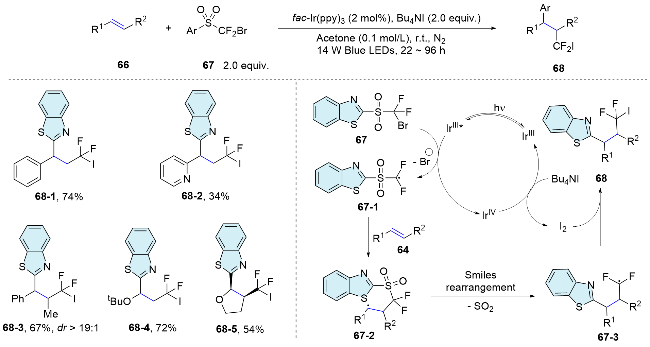
The “docking-migration” protocol was discovered by the photocatalytic difluoromethylative heteroarylation of alkenes by Zhu’s group[51] in 2018, employing a sulfone-based bifunctional reagent 67. Upon reduction by an excited-state photosensitizer, the reagent produces a difluoroalkyl radical. This radical then reacts with the alkene, resulting in the formation of a new radical species 67-2. Subsequently, a heteroaryl migration occurs via Smiles rearrangement, releasing sulfur dioxide and yielding the radical intermediate 67-3. Ultimately, interception by the in situ formed I2 leads to the formation of the desired products 68 (Scheme 24). In 2020, the research group[52] successfully achieved the arylalkylation of alkenes by using thiols as hydrogen donors. Activated and unactivated alkenes both acted as appropriate substrates, showing no sensitivity to electronic or steric influences, thereby highlighting the method extensive applicability. Moreover, a range of nitrogen-containing heteroaryls, such as thiazolyl, benzoimidazolyl, benzoxazolyl, and pyridyl, along with oxygen-containing heteroaryls like furyl and benzofuryl, and sulfur-containing heteroaryls such as thienyl and benzothienyl, as well as the oximino group, are concurrently integrated into alkenes through a sequential docking and migration process, yielding a variety of valuable products. In addition, this radical docking-migration strategy can also be applied for the difunctionallizaition of ethylene. Within a single synthetic step, the simultaneous construction of three C—C bonds ensures a rich integration of functional groups. Consequently, the simplest and most prevalent C2 feedstock ethylene is efficiently transformed into complex diheteroarylated compounds.[53]
In recent decades, significant advancements have been made in the radical-mediated difunctionalization of alkenes, however, the difunctionalization of alkynes, capable of yielding a variety of multi-substituted alkenes, has not kept pace. Taking advantage of radical rearrangement, Zhu and co-workers[54] presented a seminal study demonstrating the stereoselective transformation of aliphatic alkynes into valuable multi-substituted E-allyl alcohols via a sequence of radical migration, radical-polar crossover, and energy transfer (ET) catalyzed stereoconvergent alkene isomerization (Scheme 25).
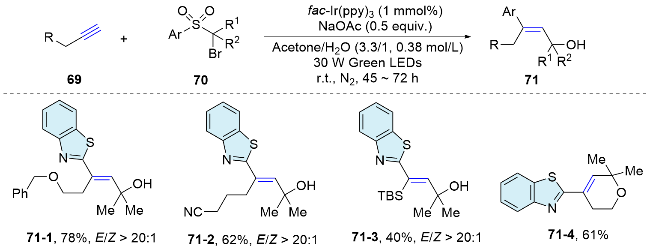
In 2018, the Stephenson group[55] described a photocatalytic method for the addition of arylsulfonyl acetamides to electron-rich alkenes, achieving anti-Markovnikov regioselective and high diastereoselective 2,2-diarylethyl-amines. In this process, single-electron oxidation of the alkene generates a radical cation species, facilitating carbon-nitrogen bond formation and yielding a key benzylic radical that undergoes a Smiles-Truce 1,5-aryl shift. This reaction is redox-neutral, demonstrates broad functional group tolerance, and proceeds at room temperature with the release of sulfur dioxide. the substrate scope, concerning both alkenes and bifunctional reagents, is constrained by the limitations of oxidation potential and activation modes (Scheme 26). In 2021, they[56] published comprehensive studies encompassing synthesis, spectroscopy, kinetics, and computation to investigate the proposed mechanism, including the pivotal aryl transfer event. These efforts to elucidate the mechanism have substantially broadened the substrate scope for the transformation, particularly concerning the migrating aryl group, and have reinforced the synthetic potential of radical aryl migrations.
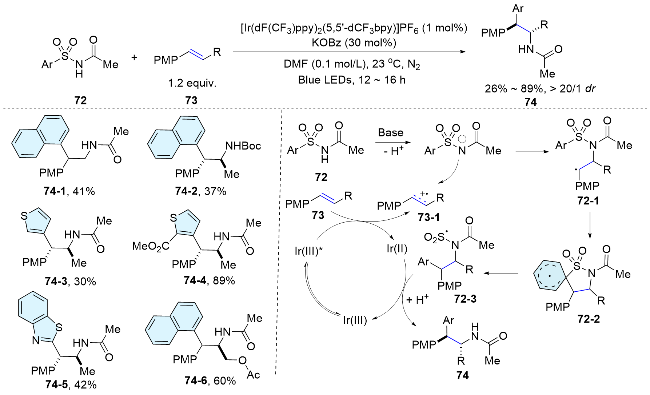
In 2024, Stephenson and colleagues[57] further expanded this bifunctional reagent mediated difunctionalization reaction. Aryl sulfinamide reagents was proved to be highly efficient for the aminoarylation of alkenes. Employing a weakly oxidizing photocatalyst, a nitrogen radical is produced under mild conditions, adding to an alkene to form a new C—N bond. Subsequently, a Smiles-Truce rearrange- ment, a desulfinylative aryl migration, occurs to establish a new C—C bond (Scheme 27). A range of unactivated alkenes underwent this type of difunctionalization reaction smoothly, showcasing the power of this radical addition/ aryl migration strategy in the field of alkene difunctionalization.
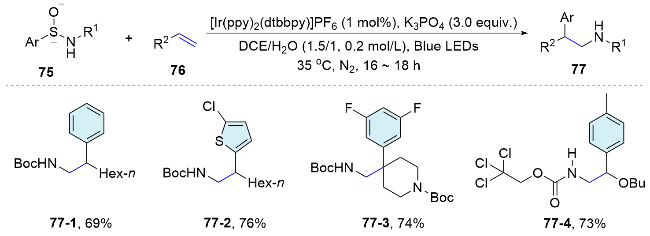
In the same year, the Nevado group[58] utilized chiral sulfoxides as linkers to connect aryl and amine groups, creating arylsulfinylamides as a bifunctional reagent for the efficient asymmetric intermolecular aminoarylation of alkenes (Scheme 28). The chiral sulfoxides acted as traceless chiral auxiliaries. Under mild photoredox conditions, the arylsulfinylamide undergoes nitrogen addition to the double bond, followed by a 1,4-aryl migration, culminating in a single operation to yield the aminoarylation adducts with high enantiomeric enrichment. A broad range of alkenes including electron-rich styrenes, aromatic vinyl amides and vinyl ethers were all amenable to this asymmetric transformation for the synthesis of optically pure β,β-diaryl- ethylamines, aryl-α,β-ethylenediamines and α-aryl-β- ami- noalcohols.
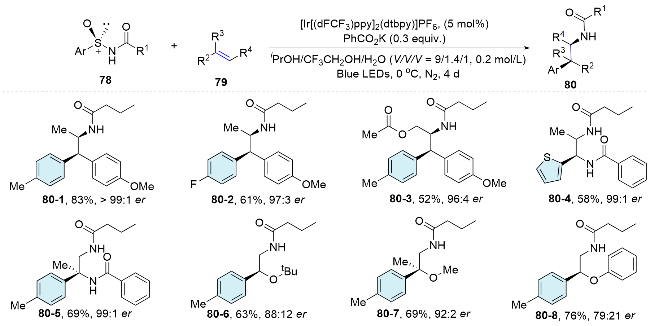
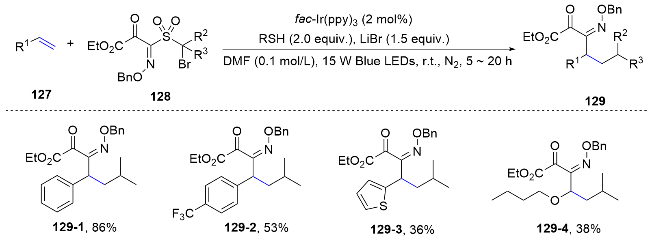
Recently, our group designed and synthesized a series of aryl sulfonylacetate derivatives as bifunctional reagents, which allowed the efficient difunctionalization of alkynes via radical addition and aryl migration cascade. Different kinds of aryl moieties from the bifunctional reagents could be efficiently introduced into both internal and terminal alkynes under mild conditions to prepare synthetically useful all carbon tetrasubstituted alkenes. To underscore the practical utility of this approach, we have successfully implemented the bifunctional reagent-mediated functionalization strategy in the synthesis of the anticancer drug Etacstil. Furthermore, the key intermediates generated by this method are also amenable to the preparation of Toremifene and Ospemifene through established cross- coupling reactions, as documented in the literature. Mechanistically, the excited photocatalyst engages in single- electron transfer with 81, producing an electrophilic alkyl radical 81-1. This radical then adds to the alkyne, forming a vinyl radical 81-2. Subsequently, it attacks the sulfonyl aryl group, leading to the formation of a spirocyclic radical species 81-3. This species undergoes homolytic cleavage of the C—S bond, releasing SO2 and generating an alkyl radical 81-4. This radical is then reduced by the photocatalyst and followed by protonation, yielding a fully carbon-substituted alkene 83 and regenerating the ground- state photocatalyst, thereby completing the photoredox catalytic cycle. Moreover, a detailed insight into the poor stereoselectivity of the obtained fully carbon-substituted alkene within this method is investigated. Under standard conditions, energy transfer from the excited photocatalyst to the generated alkenes leads to photoisomerization, yielding a mixture of cis and trans isomers (Scheme 29).[59]
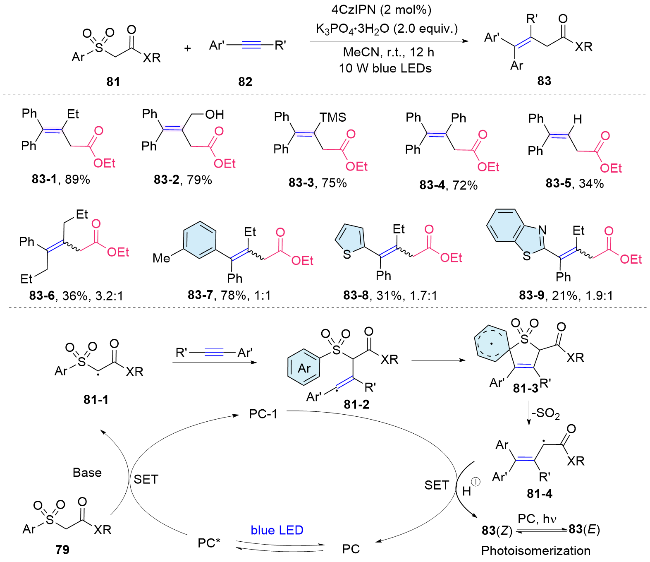
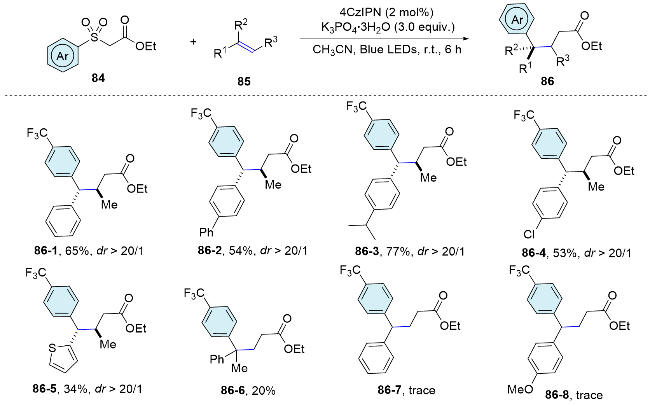
Recently, we further expended the application of this bifunctional reagent to the radical difunctionalization of alkenes. This metal-free radical process enables the simultaneous incorporation of carboxylate-bearing alkyl groups and (hetero)aryl rings into a wide range of olefins, thereby facilitating the synthesis of a diverse library of synthetically valuable γ,γ-diarylester derivatives. Complementary to the seminal works reported by Stephenson, two new carbon-carbon bonds were formed within this method. Furthermore, two activation modes have been identified, depending on the alkene substrates. For most of the substrate, a similar bifunctional reagent oxidation initiates the cascade reaction, while some of the electron-rich alkenes with relatively low oxidation potentials, the single electron oxidation of electron-rich alkenes was first involved (Scheme 30).[60]
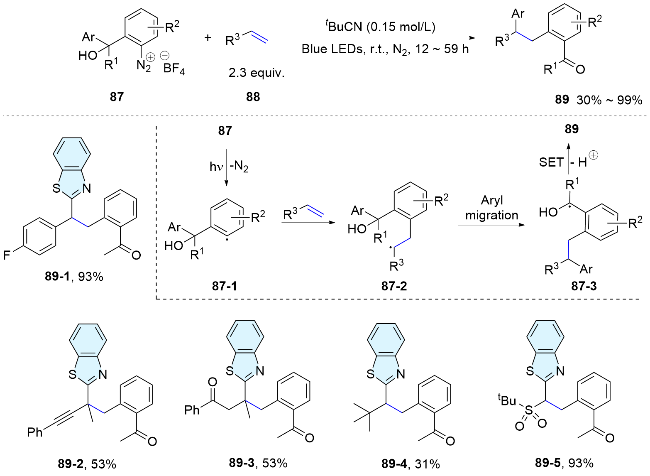
The linker in bifunctional reagents is not limited to sulfonyl group. In 2021, Zhu’s team employed functionalized aryl diazonium salts as versatile bifunctional reagents, facilitating the 1,2-diarylation of alkenes under photocatalyst free conditions. Upon light exposure, these substrates liberate nitrogen, generating aryl radicals that readily add to the alkenes, triggering Smiles rearrangement and leading to the formation of the desired products (Scheme 31).[61] Within the same year, the team[62] expanded their study by successfully utilizing aryl benzothiazolyl ether diazonium salts as effective bifunctional reagents for the diarylation of alkenes. Recently, a highly efficient method for the radical difunctionalization of aromatic alkynes has been developed by the same group, leading to the synthesis of a diverse array of valuable triarylethenes. This approach employs strategically designed aryldiazonium salts with tertiary alcohol substitution as bifunctional reagents, coupled with the use of cost-effective cuprous chloride as a catalyst. The method is characterized by its remarkable Z-selectivity and is scalable to gram-scale preparations.[63]
4 Difunctionalization via other functional group migration
In addition to aryl groups, the migrating functional groups can be expended to other unsaturated groups,[64] such as cyano groups, alkenes, alkynes, and carbonyl groups. This section focuses on recent reports concerning the difunctionalization of unsaturated hydrocarbons via radical docking-migration under visible light irradiation, categorized by the migrating groups.
4.1 Cyano group migration
In 2016, Zhu and colleagues[65] reported a groundbreaking, metal-free method for the azidocyanation of unactivated alkenes, achieving exceptional regio- and stereo- selectivity. This approach innovatively integrates an intramolecular cyano migration strategy with alkene difunctionalization for the first time, marking a significant advancement and expanding the horizons within this field. In the same year, Liu’s group[66] developed a catalytic radical method for the 1,2-cyanofunctionalization of unactivated alkenes, a process that features a remote cyano migration initiated by radical addition. Upon exposure to visible light, the excited photocatalyst can reduce sulfonyl chlorides, thereby producing electrophilic sulfinyl or fluoroalkyl radicals. These radicals then add to the alkenes, leading to the formation of a new radical species 90-1. The subsequent attack on the cyano group results in an iminyl radical intermediate, which triggers the cleavage of the C—C bond in 90-2. This cleavage facilitates the migration of the cyano group and achieves the difunctionalization of the alkene (Scheme 32).
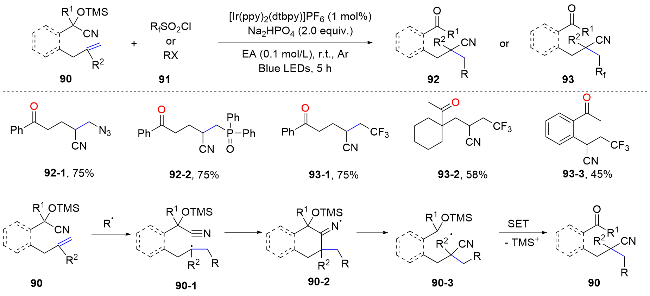
In 2017, Zhu’s team[67] leveraged distal olefinic tertiary alcohol substrates as reactive scaffolds for the cyanofluoro- alkylation of unactivated alkenes through cyano group migration. Upon exposure to visible light, fluoroalkyl radicals were generated, attacking the alkenes and initiating the cyano group migration, which led to the cyanofluoroalkylation. This method efficiently produced a wide range of synthetically valuable di- and mono-fluorinated alkyl nitriles in good yields under visible light irradiation, as depicted in Scheme 33. Taking advantage of this intramolecular cyano migration strategy, the same group successfully achieved the elusive cyanotrifluoromethylthiolation of unactivated dialkyl-substituted alkynes in a stereoselective fashion, yielding E-olefinic products. A wide range of fun- ctional groups are well tolerated under the mild reaction conditions.[68]
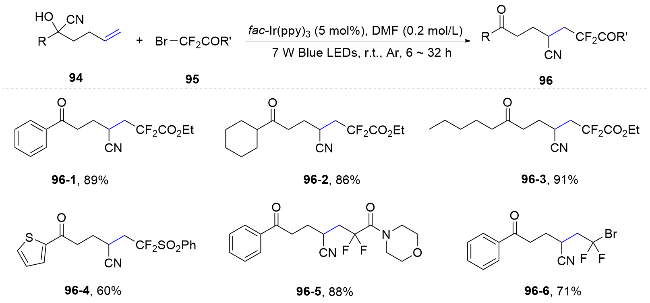
In 2022, Zhu and Chen[69] utilized distal hexenenitrile as a model substrate and introduced a novel reaction pathway characterized by functional group migration (Scheme 34). They detailed a photoredox-catalyzed trifunctionalization of 5-hexenenitriles, initiated by a CF3 radical and featuring selective cyano migration under mild conditions. The radical intermediate generated post-migration was oxidized by the photosensitizer to a carbocation, subsequently captured by an external nucleophile, yielding the final product. The compatibility of this method with various external nucleophiles allows for the introduction of remote functional groups, effectively enhancing the molecular complexity and diversity of the products.
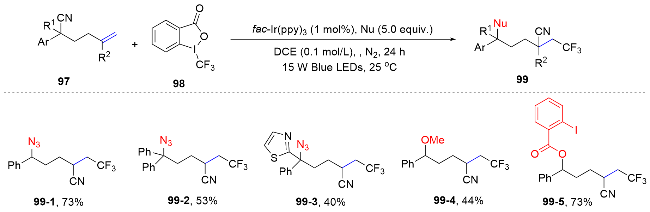
Recently, the Zhu group[70] started from the similar substrate skeleton to realize the desaturation of 5-hexene- nitriles. The sequence of reactions involves initial alkene sulfonylation, followed by an intramolecular cyano migration, and subsequent single-electron oxidation of the benzyl radical to form a cation. This is then succeeded by deprotonation, culminating in the synthesis of a diverse array of densely functionalized alkenes (Scheme 35).
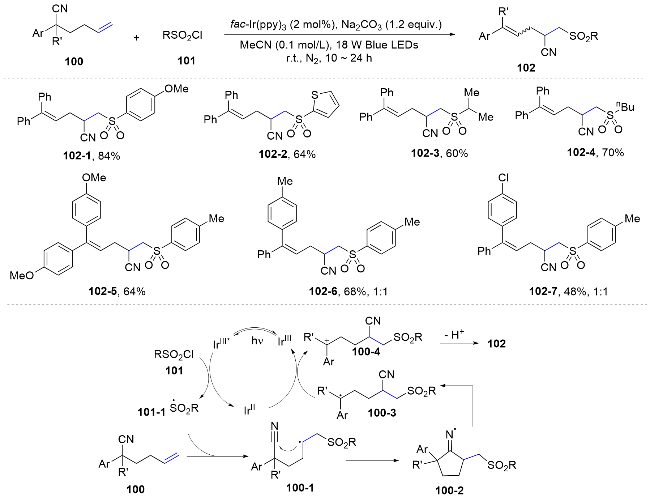
In a recent study, the same group reported an elegant cascade transformation that adeptly merges cyano migration with radical-polar crossover, thereby enabling access to a diverse array of functionalized cyclopropane derivatives from distal unactivated alkenes. The mechanistic insight reveals that the photocatalyst, upon excitation, engages in single electron oxidation to generate sulfonyl radicals. These radicals selectively add to the alkene, culminating in the formation of an alkyl radical. This intermediate undergoes intramolecular cyclization to the cyano group, resulting in a cyclic iminyl radical. Subsequent β C—C bond cleavage facilitates cyano migration, generating a new alkyl radical species. The final steps involve reduction by Ir(II) and intramolecular nucleophilic substitution, ultimately yielding the targeted cyclopropane derivatives. This innovative approach exemplifies the power of photoredox catalysis in orchestrating complex molecular rearrangements and expands the scope of cyclopropane synthesis (Scheme 36).[71]
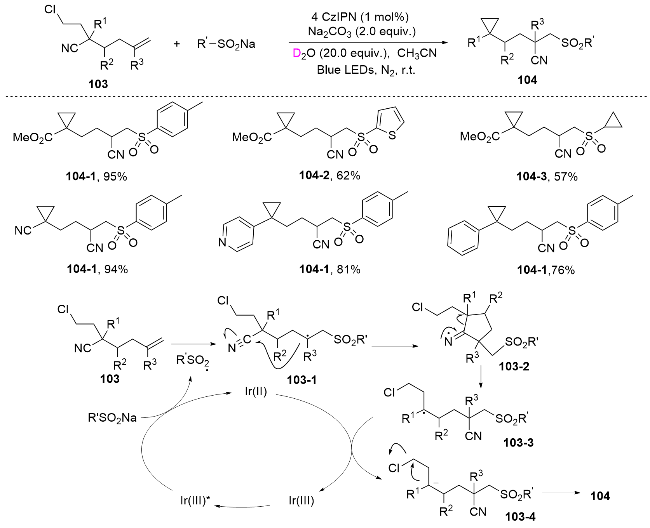
In 2023, Guo[72] employed remote alkynyl nitriles as the starting materials and successfully performed radical addition and functional group migration under photoredox catalysis conditions. Both heteroaryl groups, such as benzothiazoles, and cyano groups migrated smoothly under the standard conditions. Notably, the solvent dimethyl sulfoxide (DMSO) acted not only as a solvent but also as an oxygen source, facilitating the introduction of a remote carbonyl group (Scheme 37).
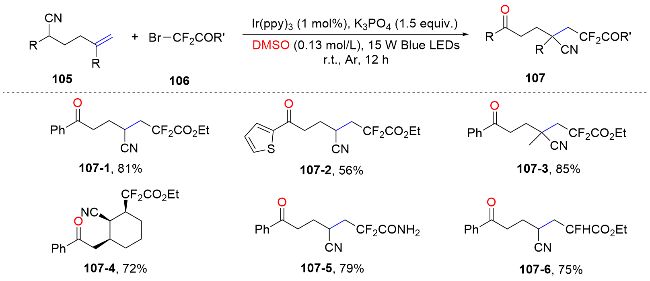
In the same year, trifluoromethyl thianthrenium salt was reported act as a precursor for trifluoromethyl radicals and as a light-absorbing reagent for the trifluoromethylcyanation of vinyl 1,1-dinitriles by Huang and co-workers.[73] Along this pathway, (Z)-alkenyl nitriles were produced via the elimination of thianthrenium ions, while aryl ketones were synthesized through a nucleophilic substitution reaction involving thianthrenium ions (Scheme 38).
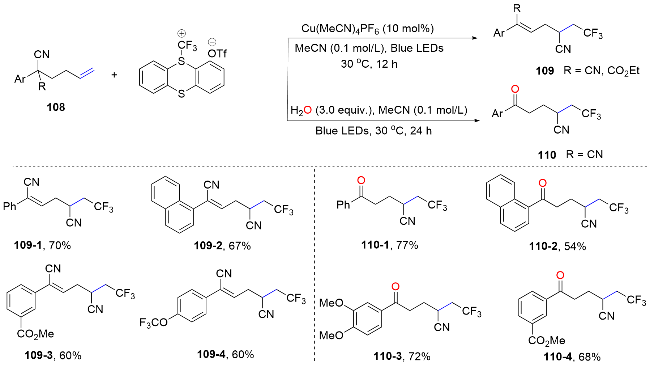
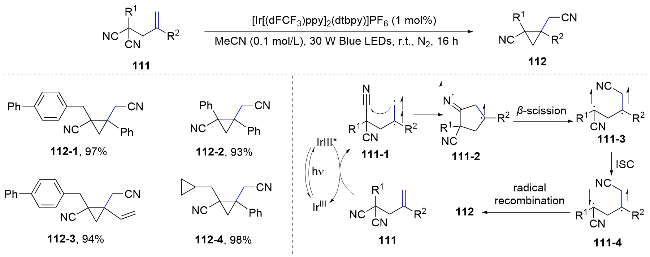
Recently, Huang’s group[74] developed a di-π-methane rearrangement reaction for the synthesis of cyclopropane derivatives via the migration of cyano functional groups to alkenes. Mechanistically, the distal di-cyano alkene substrates are excited into a diradical intermediate 111-1 through energy transfer from a triplet-state photosensitizer. Following a 5-exo-dig cyclization, an imido radical intermediate 111-2 is formed, which then undergoes β-scission to yield another diradical intermediate 111-3. Upon intersystem crossing (ISC), the newly formed diradical intermediate 111-4 may engage in a radical-radical recombination step, leading to the efficient formation of a three-membered ring.[74] This method exemplifies a broad substrate scope, atom economy, and the use of mild, redox-neutral conditions, along with the capability for late-stage modifications. The inherent limitations in classical di-π-methane rearrangements are thus addressed, leading to significant contributions to the field of synthetic methodology for the preparation of substituted cyclopropanes (Scheme 39).
4.2 Unsaturated hydrocarbon migration
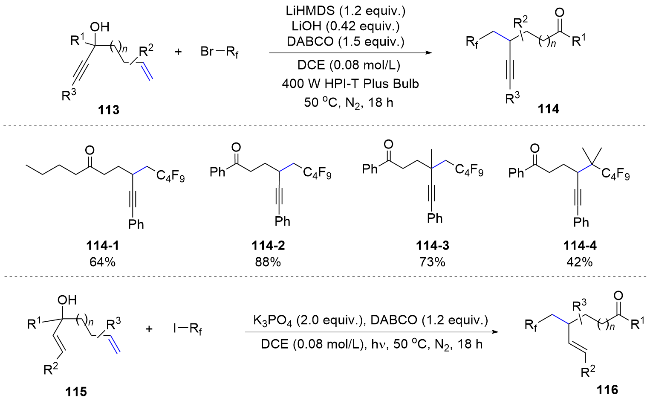
Similar to the cyano group, unsaturated hydrocarbon groups can also undergo migration in the difunctionalization reaction of distal tertiary alcohol substituted alkene substrates.[75-76] In 2017, the Studer group[77] accomplished alkyne migration by using fluoroalkyl radicals to attack alkenes with distal tertiary alcohol substituents under light irradiation, resulting in a range of alkynyl fluoroalkylated products in moderate to good yields and selectivities (Scheme 40). In the subsequent year, the group applied a similar approach to effect the migration of alkenyl groups.[78]
Furthermore, the incorporation of unsaturated hydrocarbons into bifunctional reagents provides another alternative for the difunctionalization of unsaturated hydrocarbons through radical addition and functional group migration. In 2019, Zhu’s group[79] reported a sulfonyl halide bifunctional reagent containing an alkynyl group. Under copper catalysis, the reaction achieved the alkynyl monofluoroalkylation of alkenes. Notably, after rearrangement and loss of sulfur dioxide, the resulting fluoroalkyl radical could undergo a halogen atom transfer (XAT) reaction with the solvent, allowing for the introduction of chlorine or bromine in the product by simply changing the solvent. In 2021, they[80] further introduced alkenes into the bifunctional reagent, achieving the fluoroalkylative olefination of unactivated alkenes under photocatalysis conditions and obtaining a variety of E-configured alkenes (Scheme 41, A). In the same year, the group disclosed the first example of incorporating two unsaturated carbon carbon bonds into alkenes concurrently, through photocatalytic 1,2-alkynylal-kenylation and 1,2-enynylalkenylation of alkenes (Scheme 41, B). A suite of strategically designed bifunctional reagents was designed and employed in this transformation. The sulfonyl group within these reagents stabilizes the highly reactive allenyl radical intermediate, facilitating the previously challenging intermolecular addition of the allenyl radical to alkenes. This reaction is notable for its wide applicability, as it accommodates both activated and unactivated alkenes.[81]
4.3 Carbonyl derivative migration
Introducing carbonyl compounds into tertiary alcohol substrates can also facilitate the migration of carbonyl compounds. In 2018, Zhu’s group[82] further exemplified the migration of other unsaturated C—N bond such as imines. Using imino-substituted tertiary alcohols as substrates, reactions with either difluoroalkyl or monofluoroalkyl reagents readily proceeded under visible-light irradiation, yielding the corresponding imino-migrated product 114. Both electron-rich and electron-deficient substrates were well tolerated, resulting in a series of di- and mono- fluorinated alkyl ketones. Additionally, the migration of unsaturated C—O bonds, such as the formyl group, could also be achieved under photocatalytic conditions (Scheme 42).
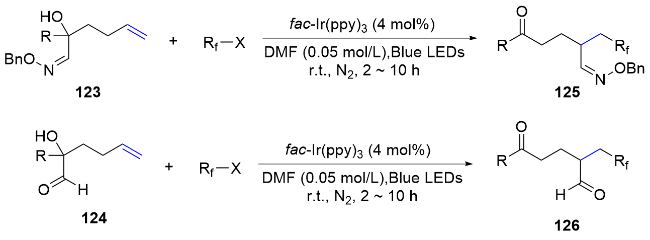
In 2020, they[52] integrated the oxime esters into a bifunctional reagent and achieved the aryl oximation of alkenes under visible light photocatalytic redox conditions (Scheme 43). In this reaction, thiols served as hydrogen donors, undergoing HAT reaction with the alkyl radicals generated after the rearrangement and loss of sulfur dioxide to form the product. The resulting sulfur radicals could engage in XAT with the substrate to activate it for participation in a radical chain cycle. In the same year, the group replaced the alkyl thiols with thioetherificating reagent, obtaining products with thioether substitution.[83]
Early in this year, Xia’s research team[84] developed a novel protocol for the 1,2-dicarbofunctionalization of una-ctivated olefins through a sequence of 1,2-acylarylations. This method leverages a photoinduced ligand-to-metal charge transfer strategy to initiate the difunctionalization, encompassing 1,2-fluoroalkylacylation, fluoroalkylarylation, and acylarylation all achieved under mild conditions. A range of (hetero)aryl, aromatic, and aliphatic acyl groups are suitable migration groups in the cascade process. And both aromatic and aliphatic aldehyde substrates can act as carbon-centered radical precursors in the 1,2-acylarylation process (Scheme 44).
4.4 Heteroatom-bearing functional group migration
In addition to the migration of functional groups containing unsaturated bonds, the migration of certain heteroatoms can also be achieved to realize the difunctionalization of unsaturated hydrocarbons. In 2020, the Studer group[85] employed fluoroalkyl radicals, produced under photoredox catalysis conditions, to react with double bonds. The nascent nucleophilic alkyl radicals then initiated the migration of the Bpin group, leading to a range of boron-containing fluoroalkylated alkenes (Scheme 45).
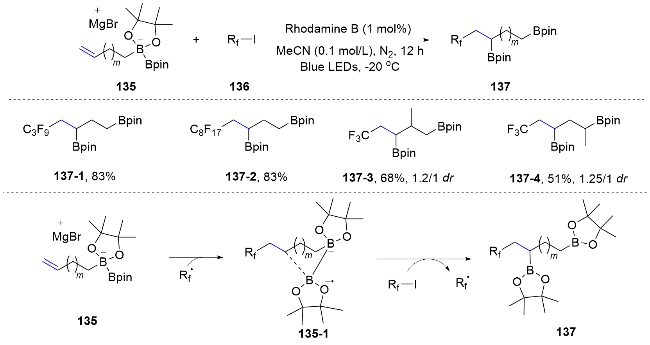
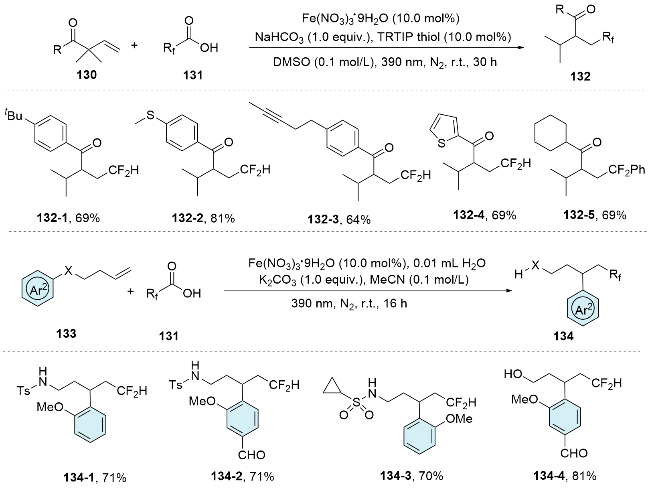
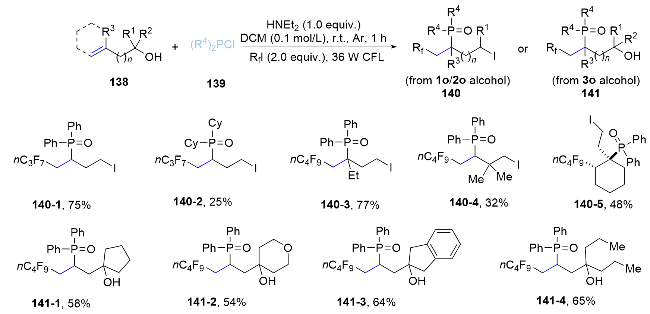
In 2022, a regioselective radical fluoroalkylphosphorylation method for unactivated alkenes through a one-pot reaction involving (bis)homoallylic alcohols, phosphine chlorides, and fluoroalkyl iodides was reported by Han and co-workers under visible light irradiation.[86] This innovative protocol features a novel radical rearrangement of alkoxyphosphine, enabling the uncommon installation of a phosphoryl group on the less sterically hindered side of terminal olefins. This method not only offers an effective approach for the regioselective introduction of both fluoroalkyl and phosphoryl groups into alkenes, but also expands the horizons of radical rearrangement chemistry (Scheme 46).
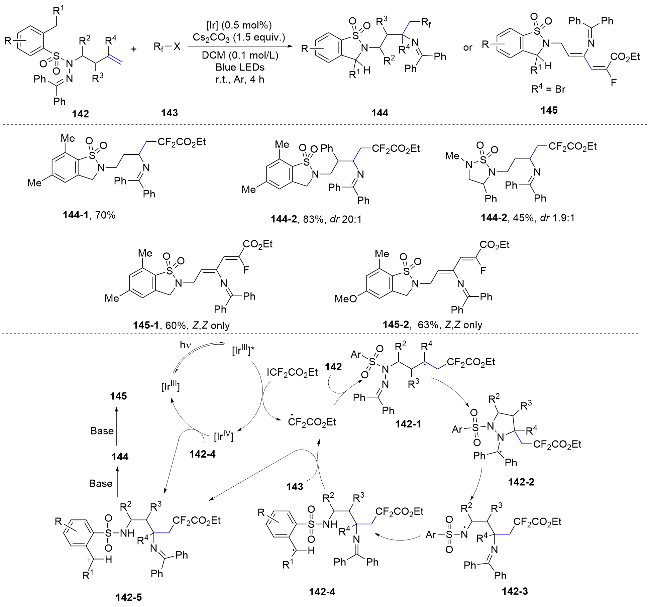
Recently, they[87] reported a method for the difunctionalization of alkenes through imino group migration. This new conversion mode enables the successful combination of alkene carboamination and Hofmann-Löffler-Freytag reaction by the reaction of N-homoallyl mesitylenesulfonyl hydrazones with ethyl difluoroiodoacetate under photoredox conditions. Mechanistically, the reaction initiates with the photoreduction of the radical precursor, which then attacks the alkene to form the carbon-centered radical 142- 1. This radical rapidly undergoes a 5-exo-trig cyclization, yielding intermediate 142-2. The cleavage of N—N bond occurs through a subsequent radical β-scission, accompanied by the generation of an amidyl radical 142-3. This radical subsequently participates in a benzylic C—H 1,5- HAT process to produce carbon-centered radical V, which is further oxidized to carbocation VI through a single- electron transfer (SET) process. Ultimately, the carbocation undergoes intramolecular nucleophilic attack by the N—H group under basic conditions. If compound 144 contains a γ-bromo group, a consecutive elimination of HBr and HF may occur due to the instability of the tertiary alkyl bromide in the presence of a base. The high photo quantum yield of the reaction suggests that a significant radical chain process may also be involved (Scheme 47).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 Conclusions and perspectives
This review provides a comprehensive perspective on photocatalyzed radical difunctionalization via functional group migration (FGM) strategy in recent decades. We categorize the representative contributions based on the specific substrate skeleton for aryl migration, bifunctional reagents mediated aryl migration and other functional group migration processes. Notably, this strategy stands out for its effective difunctionalization of alkenes and alkynes, featuring high reaction efficiency, step and atom economy, and excellent selectivity. Either specific designed distal functionalized olefin substrate or intelligently designed bifunctional reagents were all proven straightforward and efficient approach for the difunctionalization of unsaturated hydrocarbons. These methods have also provided new avenues for the concomitant formation of C—C and C—X bonds and have greatly facilitated the preparation of high-value-added products. Functional groups including aryl, heteroaryl, cyano, oximino, formyl, alkyl, alkenyl, acyl and heteroatomic substituents including an phosphorous group Bpin can all be efficiently translocated to alkene and alkynes to access the difunctionalized product. Remarkably, many seemingly impossible transformations and complex molecules that are otherwise challenging to synthesize have been achieved by means of radical rearrangements. Moreover, the underlying mechanisms of these reactions were also discussed.
Despite the notable advancements in this domain, a plethora of challenges and opportunities persist for future inquiry. The pursuit of innovative molecular design paradigms, with a special focus on the substrate framework and the role of bifunctional reagents, is deemed worthy of intensified investigated. The integration of computational chemistry and high-throughput experimental approaches to expedite the discovery and refinement of innovative molecular and catalytic systems is a critical area that requires further exploration. Moreover, the migration of heteroatoms characterized by unoccupied p- or d-orbitals, in the absence of a stabilizing π-system for radicals, presents a compelling avenue for in-depth research. There is an urgent need to devise a versatile and pragmatic methodology for enantioselective radical migration, potentially capitalizing on the power of chiral catalysts. Lastly, the strategic combination of radical addition and functional group migration to synthesize compounds with high value in the fields of pharmaceutical, organometallic, and drug development sectors continues to be a paramount goal. This pursuit underscores the enduring significance of this research area and the potential for transformative contributions to the broader scientific community.
(Lu, Y.)